- Record: found
- Abstract: found
- Article: found
Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model
Read this article at
Abstract
Background
A certain number of mutations in the Microtubule-Associated Protein Tau ( MAPT) gene have been identified in individuals with high risk to develop neurodegenerative diseases, collectively called tauopathies. The mutation A152T MAPT was recently identified in patients diagnosed with frontotemporal spectrum disorders, including Progressive Supranuclear Palsy (PSP), Frontotemporal Dementia (FTD), Corticobasal Degeneration (CBD), and Alzheimer disease (AD). The A152T MAPT mutation is unusual since it lies within the N-terminal region of Tau protein, far outside the repeat domain that is responsible for physiological Tau-microtubule interactions and pathological Tau aggregation. How A152T MAPT causes neurodegeneration remains elusive.
Results
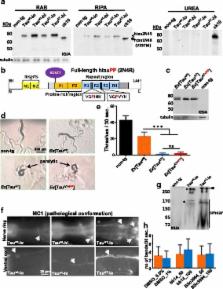
To understand the pathological consequences of this mutation, here we present a new Caenorhabditis elegans model expressing the mutant A152T MAPT in neurons. While expression of full-length wild-type human tau (Tau wt, 2N4R) in C. elegans neurons induces a progressive mild uncoordinated locomotion in a dose-dependent manner, mutant tau (Tau A152T, 2N4R) induces a severe paralysis accompanied by acute neuronal dysfunction. Mutant Tau A152T worms display morphological changes in neurons reminiscent of neuronal aging and a shortened life-span. Moreover, mutant A152T overexpressing neurons show mislocalization of pre-synaptic proteins as well as distorted mitochondrial distribution and trafficking. Strikingly, mutant tau-transgenic worms do not accumulate insoluble tau aggregates, although soluble oligomeric tau was detected. In addition, the full-length A152T-tau remains in a pathological conformation that accounts for its toxicity. Moreover, the N-terminal region of tau is not toxic per se, despite the fact that it harbours the A152T mutation, but requires the C-terminal region including the repeat domain to move into the neuronal processes in order to execute the pathology.
Conclusion
In summary, we show that the mutant Tau A152T induces neuronal dysfunction, morphological alterations in neurons akin to aging phenotype and reduced life-span independently of aggregation. This comprehensive description of the pathology due to Tau A152T opens up multiple possibilities to identify cellular targets involved in the Tau-dependent pathology for a potential therapeutic intervention.
Related collections
Most cited references58
- Record: found
- Abstract: found
- Article: not found
Kinesin superfamily motor proteins and intracellular transport.
- Record: found
- Abstract: found
- Article: not found
Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease.

- Record: found
- Abstract: found
- Article: found