- Record: found
- Abstract: found
- Article: found
Integrating whole genome sequencing, methylation, gene expression, topological associated domain information in regulatory mutation prediction: A study of follicular lymphoma

Read this article at
Graphical abstract
Abstract
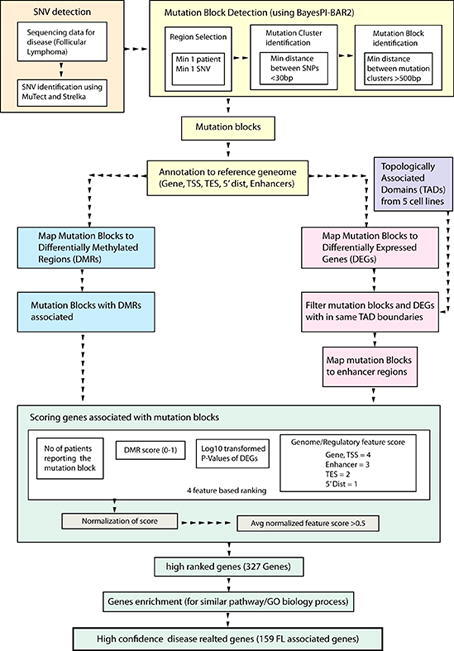
A major challenge in human genetics is of the analysis of the interplay between genetic and epigenetic factors in a multifactorial disease like cancer. Here, a novel methodology is proposed to investigate genome-wide regulatory mechanisms in cancer, as studied with the example of follicular Lymphoma (FL). In a first phase, a new machine-learning method is designed to identify Differentially Methylated Regions (DMRs) by computing six attributes. In a second phase, an integrative data analysis method is developed to study regulatory mutations in FL, by considering differential methylation information together with DNA sequence variation, differential gene expression, 3D organization of genome (e.g., topologically associated domains), and enriched biological pathways. Resulting mutation block-gene pairs are further ranked to find out the significant ones. By this approach, BCL2 and BCL6 were identified as top-ranking FL-related genes with several mutation blocks and DMRs acting on their regulatory regions. Two additional genes, CDCA4 and CTSO, were also found in top rank with significant DNA sequence variation and differential methylation in neighboring areas, pointing towards their potential use as biomarkers for FL. This work combines both genomic and epigenomic information to investigate genome-wide gene regulatory mechanisms in cancer and contribute to devising novel treatment strategies.
Related collections
Most cited references79
- Record: found
- Abstract: found
- Article: not found
featureCounts: an efficient general purpose program for assigning sequence reads to genomic features.

- Record: found
- Abstract: found
- Article: found
BEDTools: a flexible suite of utilities for comparing genomic features

- Record: found
- Abstract: found
- Article: found