- Record: found
- Abstract: found
- Article: found
Disease-specific prioritization of non-coding GWAS variants based on chromatin accessibility
Read this article at
Summary
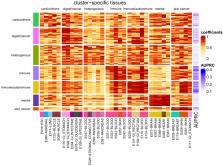
Non-protein-coding genetic variants are a major driver of the genetic risk for human disease; however, identifying which non-coding variants contribute to diseases and their mechanisms remains challenging. In silico variant prioritization methods quantify a variant’s severity, but for most methods, the specific phenotype and disease context of the prediction remain poorly defined. For example, many commonly used methods provide a single, organism-wide score for each variant, while other methods summarize a variant’s impact in certain tissues and/or cell types. Here, we propose a complementary disease-specific variant prioritization scheme, which is motivated by the observation that variants contributing to disease often operate through specific biological mechanisms. We combine tissue/cell-type-specific variant scores (e.g., GenoSkyline, FitCons2, DNA accessibility) into disease-specific scores with a logistic regression approach and apply it to ∼25,000 non-coding variants spanning 111 diseases. We show that this disease-specific aggregation significantly improves the association of common non-coding genetic variants with disease (average precision: 0.151, baseline = 0.09), compared with organism-wide scores (GenoCanyon, LINSIGHT, GWAVA, Eigen, CADD; average precision: 0.129, baseline = 0.09). Further on, disease similarities based on data-driven aggregation weights highlight meaningful disease groups, and it provides information about tissues and cell types that drive these similarities. We also show that so-learned similarities are complementary to genetic similarities as quantified by genetic correlation. Overall, our approach demonstrates the strengths of disease-specific variant prioritization, leads to improvement in non-coding variant prioritization, and enables interpretable models that link variants to disease via specific tissues and/or cell types.
Abstract
Non-coding genetic variants constitute the majority of disease-associated genetic variation in humans. In this study, Liang et al. show that variant prioritization within a specific disease context improves performance and that it enables the linking of variants to disease via specific tissues and cell types.
Related collections
Most cited references57

- Record: found
- Abstract: found
- Article: found
The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019

- Record: found
- Abstract: found
- Article: found
Integrative analysis of 111 reference human epigenomes
- Record: found
- Abstract: found
- Article: not found