- Record: found
- Abstract: found
- Article: found
Acylcarnitines : Reflecting or Inflicting Insulin Resistance?
research-article
13 December 2012
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
The incidence of obesity and insulin resistance is growing, and the increase in type
2 diabetes mellitus (DM2) constitutes one of the biggest challenges for our healthcare
systems. Many theories are proposed for the induction of insulin resistance in glucose
and lipid metabolism and its metabolic sequelae. One of these mechanisms is lipotoxicity
(1–4): excess lipid supply and subsequent lipid accumulation in insulin-sensitive
tissues such as skeletal muscle interfere with insulin-responsive metabolic pathways.
Various lipid intermediates, like ceramides, gangliosides, diacylglycerol, and other
metabolites, have been held responsible for insulin resistance (2,3,5–10). These intermediates
can exert such effects because they are signaling molecules and building blocks of
cellular membranes, which harbor the insulin receptor. In addition, lipids play an
important role in energy homeostasis. Fatty acids (FA) can be metabolized via mitochondrial
FA oxidation (FAO), which yields energy (11). As such, FAO competes with glucose oxidation
in a process known as the glucose-FA, or Randle, cycle (12).
Muoio and colleagues (1,13,14) proposed an alternative mechanism in which FAO rate
outpaces the tricarboxylic acid cycle (TCA), thereby leading to the accumulation of
intermediary metabolites such as acylcarnitines that may interfere with insulin sensitivity.
This accumulation of acylcarnitines corroborates with some human studies showing that
acylcarnitines are associated with insulin resistance (15–17). In addition, acylcarnitines
have a long history in the diagnosis and neonatal screening of FAO defects and other
inborn errors of metabolism (18). This knowledge may aid to understand the interaction
between FAO and insulin resistance and fuel future research. In this review, we discuss
the role of acylcarnitines in FAO and insulin resistance as emerging from animal and
human studies.
PHYSIOLOGICAL ROLE OF ACYLCARNITINES
Carnitine biosynthesis and regulation of tissue carnitine content.
To guarantee continuous energy supply, the human body oxidizes considerable amounts
of fat besides glucose. L-carnitine transports activated long-chain FAs from the cytosol
into the mitochondrion and is therefore essential for FAO. Carnitine is mainly absorbed
from the diet, but can be formed through biosynthesis (19). In several proteins, lysine
residues are methylated to trimethyllysine (19). Four enzymes convert trimethyllysine
into carnitine (19), of which the last step is the hydroxylation of butyrobetaine
into carnitine by γ-butyrobetaine dioxygenase (BBD). BBD is only present in human
liver, kidney, and brain, which are the sites where actual carnitine biosynthesis
takes place (19). Other tissues such as skeletal muscle must acquire carnitine from
the blood. Treatment with a synthetic peroxisome proliferator–activated receptor α
(PPARα) agonist increased BBD activity and carnitine levels in liver (20). This suggests
that the nuclear receptor PPARα, which plays a crucial role in the adaptive response
to fasting, is a regulator of (acyl)carnitine metabolism (20).
The plasmalemmal carrier OCTN2 is responsible for cellular carnitine uptake in various
organs, including reabsorption from urine in the kidney. As is the case for BBD, OCTN2
expression in liver is regulated by PPARα. A synthetic PPARα agonist increased OCTN2
expression in wild-type mice and caused a rise in carnitine levels in plasma, liver,
kidney, and heart (20). In PPARα−/− mice, low OCTN2 expression contributed to decreased
tissue and plasma carnitine levels (20).
The carnitine shuttle.
Once inside the cell, FAs are activated by esterification to CoA. Then, the carnitine
shuttle transports long-chain acyl-CoAs into mitochondria via their corresponding
carnitine ester (Fig. 1) (21). Long-chain acyl-CoAs are converted to acylcarnitines
by carnitine palmitoyltransferase 1 (CPT1), which exchanges the CoA moiety for carnitine.
CPT1 is located at the outer mitochondrial membrane, and three isoforms are known:
CPT1a, 1b, and 1c are encoded by separate genes (21). CPT1a is expressed in liver
and most other abdominal organs, as well as human fibroblasts. CPT1b is selectively
expressed in heart, skeletal muscle, adipose tissue, and testes (11). CPT1c is only
expressed in the endoplasmic reticulum (and not the mitochondria) of neurons in the
brain (22).
FIG. 1.
The carnitine shuttle. After transportation into the cell by FA transporters (FAT),
FA are activated by esterification to CoA. Subsequently, CPT1 exchanges the CoA moiety
for carnitine (C). The resulting acylcarnitine (AC) is transported across the inner
mitochondrial membrane into the mitochondrion by CACT. Once inside, CPT2 reconverts
the acylcarnitine back into free carnitine and a long-chain acyl-CoA that can undergo
FAO for ATP production via the TCA and respiratory chain (RC).
CPT1 is an important regulator of FAO flux. Glucose oxidation after a meal leads to
inhibition of CPT1 activity via the FA-biosynthetic intermediate malonyl-CoA (23),
which is produced by acetyl-CoA carboxylase (ACC) (24). There are two ACC isoforms.
ACC1 plays a role in FA biosynthesis. ACC2 has been implicated in the regulation of
FAO mainly because of its localization to the outer mitochondrial membrane (25). Conversely,
in the fasting state, activated AMP-activated protein kinase inhibits ACC resulting
in falling malonyl-CoA levels, thereby permitting CPT1 activity and thus FAO. CPT1a
is limiting for hepatic FAO and ketogenesis (26). Although the inhibition of malonyl-CoA
on CPT1b is more potent than on CPT1a, no unequivocal evidence exists showing its
control over muscle FAO (27).
FAO is also regulated at the transcriptional level. PPARα, but also PPARβ/δ, regulates
the transcription of many enzymes involved in FAO. There is ample evidence that both
PPARs participate in the transcriptional regulation of CPT1b (28–30). Regulation of
CPT1a by PPAR is less prominent (21).
After production of acylcarnitines by CPT1, the mitochondrial inner membrane transporter
carnitine acylcarnitine translocase (CACT, or SLC25A20) transports the acylcarnitines
into the mitochondrial matrix. The FA transporter CD36 possibly facilitates transfer
of acylcarnitines from CPT1 to CACT (31). Finally, the enzyme CPT2 reconverts acylcarnitines
back into free carnitine and long-chain acyl-CoAs, which can then be oxidized (21)
(Fig. 1).
Analysis of acylcarnitines.
With the introduction of tandem mass spectrometry (MS) in clinical chemistry in the
1990s, it became relatively easy to measure acylcarnitine profiles. In these profiles,
the mass-to-charge ratio reflects the length and composition of the acyl chain (32).
This technique rapidly became the preferred screening test to diagnose inherited disorders
in FAO, which lead to prominent changes in the acylcarnitine profile, with a pattern
specific for the deficient enzyme. More recently, acylcarnitine analysis is used to
investigate more common metabolic derangements such as insulin resistance.
Although most acylcarnitines are derived from FAO, they can be formed from almost
any CoA ester (18). Other intermediates that yield acylcarnitines are ketone bodies
[C4-3OH-carnitine (33)], degradation products of lysine, tryptophan, valine, leucine,
and isoleucine (C3- and C5-carnitine and others), and carbon atoms from glucose (acetylcarnitine)
(18).
The standard acylcarnitine analysis using tandem MS cannot discriminate between stereoisomers
and other isobaric compounds, which have the same nominal mass but a different molecular
structure. These compounds can be separated using liquid chromatography-tandem MS
(34). This is illustrated by C4-OH-carnitine, which can be derived from the CoA ester
of the ketone body D-3-hydroxybutyrate, (D-C4-OH-carnitine), the FAO intermediate
L-3-hydroxybutyryl-CoA (L-C4-OH-carnitine), and L-3-hydroxyisobutyryl-CoA, an intermediate
in the degradation of valine (L-isoC4-OH-carnitine) (33).
The origin of plasma acylcarnitines.
The fact that acylcarnitines can be measured in plasma illustrates that they are transported
across cell membranes. Two transporters have been implicated in the export of acylcarnitines.
In addition to import, OCTN2 can export (acyl)carnitines (35). Also, the monocarboxylate
transporter 9 (SLC16A9) may play a role in carnitine efflux (36). Although these putative
transporters have been identified, the exact nature of this transport is unknown,
but seems largely dependent on the intracellular acylcarnitine concentration (35).
Early studies in rodent heart, liver, and brain mitochondria proved mitochondrial
efflux of acylcarnitines and suggested this to be dependent on the substrate and tissue
as well as the availability of alternative acyl-CoA–utilizing reactions (37). In humans,
acylcarnitine efflux is exceptionally well-evidenced by the acylcarnitine profiles
of patients with an FAO defect (18). From a more physiological view, diets and fasting
modulate the plasma acylcarnitine profile, which reflects changes in flux through
the FAO pathway (13,16,38,39). However, exact rates of acylcarnitine production in
relation to the FAO flux under different conditions remain to be determined.
It is expected that muscle or liver contribute largely to acylcarnitine turnover.
Early studies showed that liver acylcarnitines correlated with plasma acylcarnitines
in fasted macaques, but the individual chain lengths were not studied (40). A liver–plasma
relation is plausible, considering that the liver accounts for most of the FAO activity
during fasting. Human data are lacking, but muscle acylcarnitines did not correlate
with plasma acylcarnitines during short-term fasting (16).
The physiological role of acylcarnitine efflux to the plasma compartment is unknown,
but several scenarios are likely. Acylcarnitine formation prevents CoA trapping, allowing
continuation of CoA-dependent metabolic processes (21,41). In addition to plasma,
acylcarnitines are found also in bile and urine (42,43), suggesting that acylcarnitine
efflux may serve as a detoxification process. Combined, the total daily bile and urine
production of acylcarnitine is <200 μmol. This can be calculated to be <0.01% of daily
energy requirements, which is a negligible amount in terms of potential energy loss.
Moreover, intestinal reuptake of bile acylcarnitines is possible. Alternatively, plasma
acylcarnitines may serve as a means of transportation between cells or organs or sink
for cellular/tissue acylcarnitine sequestration. Questions that remain are the contribution
of specific tissues and organs to plasma acylcarnitine levels and the turnover rates
of the individual acylcarnitine species in plasma.
ACYLCARNITINE METABOLISM IN RELATION TO INSULIN RESISTANCE
Current views on lipid metabolism in insulin resistance.
FAO may be quantitatively and qualitatively different in insulin-resistant subjects
compared with healthy subjects, but a more pertinent conundrum is if increased FAO
is either capable to limit insulin resistance via decreasing lipid accumulation or
increasing insulin resistance via accumulation of incomplete FAO products such as
acylcarnitines (1–3,13,14). Several theories describe mechanisms within the cytosol
that can cause insulin resistance (Fig. 2). It has generally been accepted that chronic
overnutrition leads to increased cytosolic lipid content of insulin-responsive tissues
(such as liver and skeletal muscle). This negatively affects the insulin sensitivity
of these tissues by inhibiting insulin signaling via intermediates as ceramide, diacylglycerol,
gangliosides, and possible other long-chain FA-derived metabolites (1,3,5–8,44). Although
contested now, cytosolic lipid accumulation was also suggested to arise from mitochondrial
dysfunction and, as a consequence, decreased FAO rate (2,9,14,45,46). Likewise, increased
levels of malonyl-CoA were suggested to limit the mitochondrial entrance of long-chain
FAs by blocking CPT1, thus resulting in accumulating cytosolic long-chain FAs and
decreasing FAO rate (10).
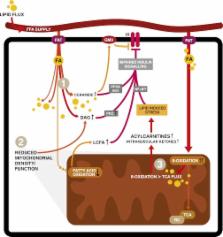
FIG. 2.
Mechanisms of lipid-induced insulin resistance. After transportation into the cell,
FA can be stored, oxidized, or used as building blocks and signaling molecules (not
all shown). Excess lipid supply and subsequent accumulation in insulin-sensitive tissues
such as skeletal muscle is proposed to interfere with different insulin-responsive
metabolic pathways via various mechanisms. Firstly (1), increased intracellular lipid
content inhibits insulin signaling via lipid intermediates such as ceramides, diacylglycerol
(DAG), or gangliosides (GM3) via effects on protein phosphatase A2 (PPA2) and protein
kinase B (Akt), protein kinase C (PKC), or effects on the insulin receptor in the
cell membrane (1,3,5–8,44). Effects of lipid intermediates on inhibitors of nuclear
factor-κβ (NFκB) kinase subunit β and c-Jun N-terminal kinase 1 are not depicted.
The second mechanism (2) is a decreased number of functional mitochondria resulting
in lower FAO rates and increased accumulation of cytosolic lipid, again interfering
with insulin sensitivity (2,9). Finally (3), metabolic overload of mitochondria leads
to incomplete β-oxidation. In this figure, oxidation of FA outpaces the TCA and respiratory
chain (RC), resulting in intramitochondrial accumulation of FAO intermediates like
acylcarnitines. These subsequently impinge on insulin signaling (1,48,50–56). In this
figure, only the direct effects of acylcarnitines on nuclear factor-κβ have been proposed
(70).
Alternatively, more recent mechanistic (13,47,48) and metabolomic (49–54) studies
associated obesity-induced insulin resistance with intramitochondrial disturbances.
In this model, lipid overload leads to increased rather than decreased FAO in skeletal
muscle. This coincides with accumulating acylcarnitines, an inability to switch to
carbohydrate substrate, and a depletion of TCA intermediates, suggesting that FAO
flux does not match TCA flux, leading to incomplete FAO (13,47,48). In vitro interfering
with FA uptake in L6 myocytes or a coordinate induction of FAO and TCA enzymes by
exercise or PPARγ coactivator 1α overexpression prevented insulin resistance (13,48).
Moreover, using carnitine to stimulate FAO without affecting the TCA in these myocytes
was dose-dependently associated with insulin resistance (13). Zucker Diabetic Fatty
rats, a model for more severe insulin resistance, had higher acylcarnitines but lower
TCA intermediates (such as citrate, malate, and succinate) in skeletal muscle, again
suggesting that increased FAO induces insulin resistance when not followed by proportionally
increased TCA activity (13). Additionally, the malonyl-CoA decarboxylase−/− mouse
that had decreased FAO due to higher malonyl-CoA concentrations resisted diet-induced
insulin resistance, which further implicated FAO in the pathogenesis of insulin resistance
(13). The available studies on acylcarnitine metabolism and the relationship with
insulin resistance will be discussed in the next sections with a focus on human studies.
The effect of increased lipid flux on mitochondrial FA uptake and oxidation: implications
for insulin sensitivity.
Insulin-dependent DM2 patients had lower (∼25%) carnitine concentrations, especially
with longer-standing or complicated disease (55,56). Interestingly, carnitine infusions
increased FAO in lean healthy subjects, but only when high-dose insulin was coadministered
(57,58), which may be explained by an increased muscle OCTN2 expression under these
conditions (59). The importance of insulin for cellular carnitine uptake is underscored
by the finding that insulin and carnitine administration lowered muscle malonyl-CoA
and lactate concentrations, whereas muscle glycogen increased (58). These findings
are supported by animal studies, which demonstrated that carnitine levels were diminished
in skeletal muscle of multiple insulin-resistant rat models. A high-fat diet (HFD)
exacerbated the age-related decrease of tissue carnitine content in these rats (primarily
skeletal muscle, liver, and kidney) (60). Moreover, carnitine supplementation of HFD
animals decreased plasma glucose levels and homeostasis model assessment indices (60,61).
Likewise, carnitine supplementation improved insulin-stimulated glucose disposal in
mouse models of diet-induced obesity and genetic diabetes (62). Recently, it was shown
that 6 months of carnitine supplementation improved glucose homeostasis in insulin-resistant
humans (14).
Although supplementation of carnitine possibly augments FAO and insulin sensitivity,
the lower carnitine levels in diabetes patients are unexplained. On the one hand,
carnitine uptake is insulin-dependent and therefore the absence of or resistance to
insulin may be the cause of lower carnitine levels. On the other hand, higher lipid
load may lead to higher acylcarnitine concentrations and thus lower free carnitine.
In addition, several studies reported on the carnitine shuttle and its effects on
the rate of FAO in the development of insulin resistance. Obese subjects had lower
CPT1 and citrate synthase content in muscle and lower FAO, suggesting that lesions
at CPT1 and post-CPT1 events (i.e., mitochondrial content) may lower FAO in obesity
(63). Although short-term inhibition of CPT1 with etomoxir in humans did not impede
insulin sensitivity despite increased intramyocellular lipid accumulation (64), prolonged
inhibition in rats resulted in the accumulation of intramyocellular lipid and increased
insulin resistance while doubling adiposity despite feeding a low-fat diet (65). These
results all led to the assumption that low FAO rates due to decreased function of
CPT1 were associated with insulin resistance, possibly caused by an accumulation of
intramyocellular lipid intermediates and their interference with insulin signaling.
Indeed, CPT1 activity increased after an endurance training program in obese subjects,
coinciding with increased FAO, improved glucose tolerance, and insulin sensitivity
(66). However, this may also be explained by the stimulatory effect of endurance training
on mitochondrial function (i.e., TCA and respiratory chain activity), thereby relieving
the heavy lipid burden on mitochondria (48,67). In contrast to the model in which
excess FAO induces insulin resistance, these data suggest that decreasing mitochondrial
FA uptake results in elevated intramuscular lipid levels and subsequent insulin resistance.
However, increasing FAO by carnitine treatment in animals and humans permits mitochondrial
FA uptake and oxidation that benefits insulin sensitivity. These observations will
have to be reconciled with other studies that implicated incomplete FAO and acylcarnitine
accumulation in the pathogenesis of insulin resistance.
Short-chain acylcarnitines in insulin resistance.
Older work reported elevated acylcarnitine levels in obese insulin-resistant subjects
(15), but acylcarnitines were not suggested to be implicated in insulin resistance
at that time. The shortest acylcarnitine, acetylcarnitine, is of particular interest
because it may illustrate the controlling role of acetyl-CoA on substrate switching
and thus metabolic flexibility. The mitochondrial enzyme carnitine acetyl-CoA transferase
(CrAT) converts acetyl-CoA to the membrane-permeable acetylcarnitine and permits mitochondrial
efflux of excess acetyl-CoA that otherwise could inhibit pyruvate dehydrogenase (68).
Infusing intralipid decreased insulin sensitivity while increasing muscle acetylcarnitine
(69). The same was true for plasma and muscle acetylcarnitine levels under high FAO
conditions (starving), suggesting upregulation of CrAT to traffic acetyl-moieties
(16). In contrast to lower CrAT expression in diabetic subjects (68), plasma acetylcarnitine
levels showed significant positive correlation with HbA1c levels over a wide range
of insulin sensitivity, suggesting upregulation of CrAT in insulin-resistant states
(70).
There is some complexity, as both lipid and glucose oxidation funnel into acetylcarnitine
as supported by different findings (68,71). First, the insulin-mediated suppression
of muscle acetylcarnitine occurred under high FAO conditions, but not postabsorptively
(i.e., higher glucose availability) (16). Also, muscle acetylcarnitine correlated
negatively with FAO in the postabsorptive state (71), whereas plasma acetylcarnitine
correlated with plasma glucose levels in the postprandial state (72). In light of
these data, the question is interesting if CrAT really favors FA-derived acetyl-CoA
over glucose-derived acetyl-CoA because this might imply intracellular compartmentalization
of acetyl-CoA (68). Moreover, glucose-derived acetyl-CoA can be carboxylated by ACC,
producing the CPT1 inhibitor malonyl-CoA. Direct effects of FAO-derived acetyl-CoA
on insulin action are unknown.
C4-OH-carnitine (i.e., the carnitine ester of 3-hydroxybutyrate) has been proposed
to cause insulin resistance: hepatic overexpression of malonyl-CoA decarboxylase in
rats on an HFD reversed whole-body, liver, and muscle insulin resistance while only
decreasing C4-OH-carnitine within the acylcarnitine profile (47). In fasted humans,
plasma and muscle C4-OH-carnitine increased (33). The increase in C4-OH-carnitine
in these animal and human studies is quantitatively much more pronounced then the
increase in acetylcarnitine; thus, C4-OH-carnitine production may exert greater demands
on cellular carnitine stores. Moreover, ketone bodies yield acetyl-CoA, which stimulates
PDK4 and thus inhibits glucose oxidation (73). In summary, under conditions characterized
by higher FAO, elevated short-chain acylcarnitines may reflect higher lipid fluxes,
but a direct relation to insulin resistance remains to be established.
Amino acid–derived acylcarnitines in insulin resistance.
Metabolomics showed that branched-chain and aromatic amino acids (isoleucine, leucine,
valine, tyrosine, and phenylalanine) (74) significantly correlated with present or
future diabetes (54,74,75). In line with this, the branched-chain amino acid–derived
C3- and C5-carnitine, together with FA-derived C6- and C8-carnitine, were higher in
obese and DM2 subjects compared with lean controls (17,54). In the same study, C4-dicarboxylcarnitine
(C4DC-carnitine), also derived from branched-chain amino acid metabolism, showed a
positive correlation with basal glucose levels and HbA1c (17). In comparison with
obese non–insulin-resistant subjects, DM2 subjects also had higher C3- and C5-carnitine
levels compared with controls during insulin administration. In this study, C3- but
not C5-carnitine correlated negatively with glucose disposal (17).
At first glance, correlations of acylcarnitines to surrogate markers of insulin resistance
fit with mitochondrial overload and incomplete FAO. Acylcarnitines, however, also
directly reflect the oxidation rate of FA and amino acids, which is supported by human
nutritional intervention studies (16,33,38,39). The uncertainty regarding the direct
interference of short-chain acylcarnitines and their metabolism with insulin-signaling
processes and insulin sensitivity warrants care when attributing a primary role for
amino acid–derived acylcarnitines in the induction of insulin resistance.
Medium- and long-chain acylcarnitines: more evidence for insulin-resistant effects?
Long-chain FA such as palmitic acid were associated with insulin resistance, making
a role for long-chain acylcarnitines such as C16 in insulin resistance conceivable
(3,44). In 1980, Hoppel et al. (15) showed that the fasting-induced increase in plasma
acylcarnitines was restored upon refeeding in lean subjects within 24 h opposed to
4 days in obese subjects, suggesting an impaired metabolic flexibility in the latter.
The hypothesis that obesity-induced alteration in the acylcarnitine profile are caused
by incomplete FAO was based largely on two animal studies by the same group showing
that long-chain acylcarnitine species (C16, C18:2, C18:1, and C18) were persistently
increased in diet-induced obese rats, in both the fed and fasted state (13,48). As
reported for humans, most acylcarnitine species decreased upon refeeding in the chow-fed
control group, but not in the obese animals, suggesting they were incapable of adjusting
their metabolism in response to refeeding. Although excessive and incomplete FAO can
be responsible for insulin resistance, it can be argued that FAO probably must be
in relative excess to oxidation in TCA and respiratory chain in order to guarantee
continuous energy supply.
Obese and insulin-resistant humans had higher plasma long-chain acylcarnitine levels
compared with lean controls (17). Upon insulin infusion, long-chain acylcarnitines
decreased overall, but to a lesser degree in the diabetic subjects. This was in agreement
with lower resting energy expenditure, indicating ongoing FAO or lipid flux (metabolic
inflexibility) (17). Moderate correlations between acylcarnitine profiles and various
clinical characteristics (i.e., higher BMI, basal free FA levels, insulin sensitivity)
point at a causal relationship. The DM2 subjects were unable to suppress acylcarnitines
during insulin infusion in contrast to matched obese controls; therefore, elevated
long-chain acylcarnitines in the diabetic group likely reflect increased lipid flux
and illustrate the tight connection of acylcarnitines with FAO flux (17).
Postprandially, plasma long-chain acylcarnitines did decrease in obese insulin-resistant
subjects, but the magnitude of this decrease correlated with both premeal insulin-mediated
glucose disposal rates and FAO and has been largely explained by nadir levels of C12:1,
C14, and C14:1-carnitine (72). This showed that the more insulin-sensitive subjects
are, the more capable they are at metabolizing FAs. Metabolomics in healthy, overweight,
calorie-restricted subjects yielded comparable results; in this study, acylcarnitines
correlated significantly with plasma insulin and free FA levels, albeit with low correlation
coefficient (49).
All in all, acylcarnitines with longer chain lengths are associated with insulin resistance,
which seems logic in the light of known effects of long-chain FAs on insulin signaling.
Indeed, acylcarnitines can reside in cell membranes because they are amphipathic molecules.
Increasing chain length favors partitioning into the membrane phase (e.g., C16- and
18-carnitine) (76). It is interesting to speculate that long-chain acylcarnitines
can interfere with insulin signaling directly within the cell membrane (3). In contrast,
acylcarnitines seem to track with higher lipid flux and as such may only indicate
higher FAO.
ACYLCARNITINES: REFLECTING OR INFLICTING INSULIN RESISTANCE?
The concept of lipotoxicity is generally accepted in the field of obesity-induced
impairment of insulin sensitivity, and more and more attention has attributed to intramitochondrial
alterations and impairments in FAO, thereby focusing on acylcarnitines (1). Collected
evidence shows that acylcarnitines have distinct functions in mitochondrial lipid
metabolism. The transmembrane export of acylcarnitines suggests that they not only
prevent the accumulation of noxious acyl-CoAs, but also reduce CoA trapping, which
is crucial for many metabolic pathways (21,41). Additionally, the metabolism of short-chain
acylcarnitines and the interaction of acetyl-CoA and acetylcarnitine via CrAT may
regulate the pyruvate dehydrogenase complex, thereby affecting glucose oxidation (68).
Besides mitochondrial need to liberate CoA and export acetyl-CoA, acylcarnitines may
simply reflect the FAO flux.
The concept of increased, though incomplete, FAO by disproportional regulation of
FAO, TCA, and respiratory chain is attractive to explain insulin resistance. However,
there remains doubt about this mechanism, and there is no proof that acylcarnitines
play a role in the induction of insulin resistance itself. Acylcarnitines are present
under physiological conditions, and their levels vary according to dietary circumstances
(13,16,38,39). The acylcarnitine fluxes are unknown but probably much lower than FAO
flux. Moreover, it can be argued that flux of FAO probably will be in relative excess
to downstream oxidation in TCA and respiratory chain to guarantee continuous substrate
supply and allow fine tuning and anticipation for metabolic changes (e.g., activity).
Otherwise, the organism’s response to increased energy demands will be attenuated,
leading to more severe impairment of mitochondrial function as evidenced by the inherited
FAO disorders.
Observational studies associating different acylcarnitines to a variety of end points
may yield new hypotheses but are unlikely to move the field forward from a mechanistic
perspective. Many questions are unanswered, and some issues deserve particular attention.
Tracer studies can quantify FAO flux and acylcarnitine production in different insulin-resistant
models on the cellular, tissue, and whole-organism level. Multiple animal and human
models can help to investigate the effect of carnitine availability on insulin sensitivity.
Mouse models for and humans with primary carnitine deficiency can be used to investigate
the effect of carnitine availability on substrate switching and insulin sensitivity.
In vitro work in muscle or liver cell lines is still important to dissect the influence
of acylcarnitines on conventional insulin signaling or mechanisms of nutrient-induced
mitochondrial stress. In this respect, different animal and human FAO disorders that
accumulate acylcarnitines may undergo insulin sensitivity testing. The contribution
of different organs to plasma acylcarnitines can be investigated using transorgan
arteriovenous balance isotope-dilution techniques under different conditions. Finally,
we may set foot in new areas in which acylcarnitines may have unexpected roles, like
interaction with the insulin receptor in the plasma membrane or signaling in the gut
when cosecreted with bile. Recently, magnetic resonance spectroscopy was shown to
image tissue acetylcarnitine in humans enabling noninvasive techniques to assay tissue
acetylcarnitine (77). All of these studies and more are necessary to decide to what
extent acylcarnitines are reflecting or inflicting insulin resistance.