- Record: found
- Abstract: found
- Article: found
High-Resolution Pictures of AML Hierarchies
review-article
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Clonal heterogeneity is believed to be a cancer hallmark. This is best exemplified
by acute myeloid leukemia (AML), an aggressive hematopoietic malignancy in which myeloid
progenitors accumulate in the bone marrow. Primary AML tumors contain multiple subclones,
which display distinct sets of cytogenetic abnormalities, somatic mutations, epigenetic
features, and functional properties.
1
This multifaceted heterogeneity, moreover, is dynamic as the clonal composition of
the tumor evolves during disease progression and relapse.
Although single-cell genomic technologies
2
have greatly improved the characterization of AML biology,
3
they present several shortcomings. Standard single-cell RNA sequencing (scRNAseq)
methods are able to read full-length transcripts but they lack sufficient throughput
to discern malignant from normal cells. Conversely, digital technologies, such as
nanowell-based scRNAseq, which provide higher-resolution data, are not able to fully
capture the mutational status of malignant cells as they present a 3’ bias in the
read coverage.
In a recent issue of Cell, Peter van Galen and colleagues moved the technology one
step forward and investigated AML hierarchies performing both transcriptional and
mutational analysis at the single-cell level
4
(Fig. 1). The authors profiled 38,410 single cells from 16 AML patients and 5 normal
bone marrow aspirates using a high-throughput nanowell-based scRNAseq (seq-well),
which they adapted to sequence frequently mutated AML genes. To do so, the researchers
took advantage of an amplification step in the transcriptome protocol, which generated
full-length cDNAs bearing cell-specific barcodes appended to the 3’ ends. Using primers
adjacent to the mutational sites previously detected by targeted DNA sequencing, they
next generated amplicons containing mutational sites and barcodes. Sequencing these
amplicons by short- and long-read sequencing provided comprehensive genotyping of
individual cells (ie, insertions, deletions, fusions, and point mutations of recurrently
mutated AML genes). Transcriptomic and genotyping data were than integrated using
a machine learning algorithm to distinguish malignant from normal cells.
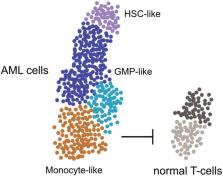
Figure 1
Clonal heterogeneity of AML. Single-cell transcriptomic and mutational analysis revealed
that AML samples have a variable cell-type composition, which correlates with genetics,
surface markers, cellular morphology, and patient outcome. Less differentiated cells
had stem cell characteristics, while more differentiated myeloid cells were shown
to have an immunosuppressive function negatively affecting normal T-cells.
The massive amount of data thereby generated was next interrogated to elucidate the
composition of cellular hierarchies. To this end, the researchers first classified
the leukemic cells based on their similarity to their normal bone marrow counterparts.
This analysis identified 6 malignant AML cell types (hematopoietic stem cell (HSC)-like,
progenitor-like, granulocyte-macrophage progenitor (GMP)-like, promonocyte-like, monocyte-like
and conventional dendritic cell-like), whose relative abundance markedly varied between
patient samples and correlated with the cellular morphology and surface phenotypes
of the tumor bulk as well as patient outcome. In a second step, the authors obtained
gene signatures for each of these AML cell types and used them to hierarchically cluster
the bulk expression profiles of 179 diagnostic AML samples from the cancer genome
Atlas. This strategy led the researchers to identify 7 different cellular clusters.
Most of them comprised leukemias characterized by the predominance of 1 specific cell
type (eg, progenitor-like), while another cluster included leukemias containing several
malignant cell types along the differentiation spectrum (ie, from the hematopoietic
stem cell-like to the myeloid-like type). Interestingly, each of these cell composition-based
clusters closely correlated with prototypic genetic lesions, thus suggesting that
genetics is an important force shaping the cellular composition in AML.
Lastly, the authors investigated in more depth 2 cell types at the opposite ends of
the differentiation spectrum, namely the HSC-like AML cells and the monocyte-like
cells. Confirming previous studies,
5
HSC-like cells were found to co-express stemness-related and myeloid-priming genes.
Monocyte-like cells, instead, expressed immunomodulatory factors and immunosuppressive
myeloid markers, and strongly inhibited T-cell activation in vitro (Fig. 1). Albeit
variable in abundance, myeloid-like cells were found in most of the AML samples analyzed,
thus suggesting that they may play important roles in shaping an immunosuppressive
microenvironment in the bone marrow. Functional studies will be necessary to extend
these observations and dissect the mechanisms by which myeloid-like AML cells contribute
to the development of the disease. Although it remains under debate whether T-cells
can interact with and eliminate leukemia stem cells (LSCs), it will be intriguing
to explore this scenario and verify whether the myeloid-like AML cells protect LSCs
from immune-mediated elimination. Along this line and supporting previous findings,
6
T-regulatory cells, a subset of T-cells endowed with immunosuppressive properties,
were decreased in bone marrow aspirates from AML patients. In light of these findings,
it is worth dissecting how myeloid-like AML cells affect the leukemic bone marrow
microenvironement and LSC niches. Last, but not least, it will be important to exploit
the technological advances developed by van Galen and colleagues to characterize preleukemic
clones as well as the heterogeneity at the LSC level.
Related collections
Most cited references2
- Record: found
- Abstract: found
- Article: not found
Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation?
Celalettin Ustun, Jeffrey S. Miller, David Munn … (2011)
- Record: found
- Abstract: not found
- Article: not found
Single-cell sequencing in normal and malignant hematopoiesis
NK Wilson, B. Göttgens (2018)