- Record: found
- Abstract: found
- Article: found
Deciphering functional redundancy in the human microbiome
Read this article at
Abstract
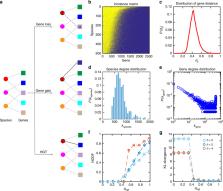
Although the taxonomic composition of the human microbiome varies tremendously across individuals, its gene composition or functional capacity is highly conserved — implying an ecological property known as functional redundancy. Such functional redundancy has been hypothesized to underlie the stability and resilience of the human microbiome, but this hypothesis has never been quantitatively tested. The origin of functional redundancy is still elusive. Here, we investigate the basis for functional redundancy in the human microbiome by analyzing its genomic content network — a bipartite graph that links microbes to the genes in their genomes. We find that this network exhibits several topological features that favor high functional redundancy. Furthermore, we develop a simple genome evolution model to generate genomic content network, finding that moderate selection pressure and high horizontal gene transfer rate are necessary to generate genomic content networks with key topological features that favor high functional redundancy. Finally, we analyze data from two published studies of fecal microbiota transplantation (FMT), finding that high functional redundancy of the recipient’s pre-FMT microbiota raises barriers to donor microbiota engraftment. This work elucidates the potential ecological and evolutionary processes that create and maintain functional redundancy in the human microbiome and contribute to its resilience.
Abstract
Here, the authors develop a genome evolution model to investigate the origin of functional redundancy in the human microbiome by analyzing its genomic content network and illustrate potential ecological and evolutionary processes that may contribute to its resilience.
Related collections
Most cited references64

- Record: found
- Abstract: found
- Article: found
Structure, Function and Diversity of the Healthy Human Microbiome
- Record: found
- Abstract: found
- Article: not found
A human gut microbial gene catalogue established by metagenomic sequencing.
- Record: found
- Abstract: found
- Article: not found