- Record: found
- Abstract: found
- Article: found
The PUF binding landscape in metazoan germ cells
Read this article at
Abstract
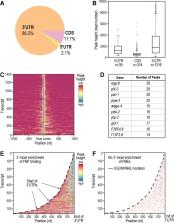
PUF ( Pumilio/ FBF) proteins are RNA-binding proteins and conserved stem cell regulators. The Caenorhabditis elegans PUF proteins FBF-1 and FBF-2 (collectively FBF) regulate mRNAs in germ cells. Without FBF, adult germlines lose all stem cells. A major gap in our understanding of PUF proteins, including FBF, is a global view of their binding sites in their native context (i.e., their “binding landscape”). To understand the interactions underlying FBF function, we used iCLIP (individual-nucleotide resolution UV crosslinking and immunoprecipitation) to determine binding landscapes of C. elegans FBF-1 and FBF-2 in the germline tissue of intact animals. Multiple iCLIP peak-calling methods were compared to maximize identification of both established FBF binding sites and positive control target mRNAs in our iCLIP data. We discovered that FBF-1 and FBF-2 bind to RNAs through canonical as well as alternate motifs. We also analyzed crosslinking-induced mutations to map binding sites precisely and to identify key nucleotides that may be critical for FBF–RNA interactions. FBF-1 and FBF-2 can bind sites in the 5′UTR, coding region, or 3′UTR, but have a strong bias for the 3′ end of transcripts. FBF-1 and FBF-2 have strongly overlapping target profiles, including mRNAs and noncoding RNAs. From a statistically robust list of 1404 common FBF targets, 847 were previously unknown, 154 were related to cell cycle regulation, three were lincRNAs, and 335 were shared with the human PUF protein PUM2.
Related collections
Most cited references94
- Record: found
- Abstract: found
- Article: not found
Fast gapped-read alignment with Bowtie 2.
- Record: found
- Abstract: found
- Article: not found
Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources.

- Record: found
- Abstract: found
- Article: found