- Record: found
- Abstract: found
- Article: found
Physics-inspired machine learning of localized intensive properties†

Read this article at
Abstract
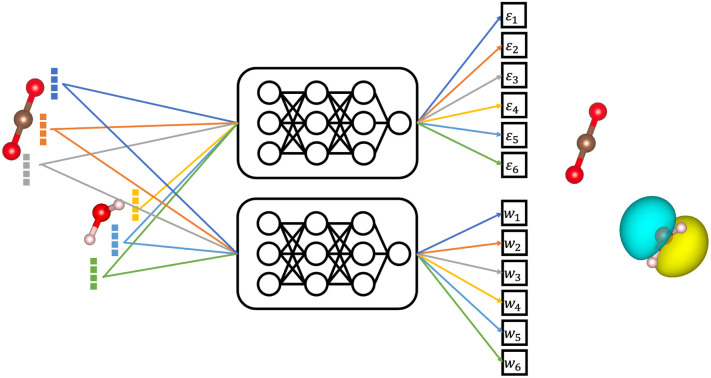
Machine learning (ML) has been widely applied to chemical property prediction, most prominently for the energies and forces in molecules and materials. The strong interest in predicting energies in particular has led to a ‘local energy’-based paradigm for modern atomistic ML models, which ensures size-extensivity and a linear scaling of computational cost with system size. However, many electronic properties (such as excitation energies or ionization energies) do not necessarily scale linearly with system size and may even be spatially localized. Using size-extensive models in these cases can lead to large errors. In this work, we explore different strategies for learning intensive and localized properties, using HOMO energies in organic molecules as a representative test case. In particular, we analyze the pooling functions that atomistic neural networks use to predict molecular properties, and suggest an orbital weighted average (OWA) approach that enables the accurate prediction of orbital energies and locations.
Abstract
A physics-inspired machine learning approach to predicting localized intensive properties in molecules is presented. The new method is applied to predicting orbital energies and localisations in potential organic semiconductors.
Related collections
Most cited references56
- Record: found
- Abstract: found
- Article: not found
Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy.
- Record: found
- Abstract: found
- Article: not found
Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections.
- Record: found
- Abstract: found
- Article: not found