- Record: found
- Abstract: found
- Article: found
High-throughput prioritization of target proteins for development of new antileishmanial compounds
Read this article at
Abstract
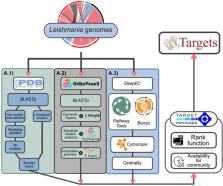
Leishmaniasis, a vector-borne disease, is caused by the infection of Leishmania spp., obligate intracellular protozoan parasites. Presently, human vaccines are unavailable, and the primary treatment relies heavily on systemic drugs, often presenting with suboptimal formulations and substantial toxicity, making new drugs a high priority for LMIC countries burdened by the disease, but a low priority in the agenda of most pharmaceutical companies due to unattractive profit margins. New ways to accelerate the discovery of new, or the repositioning of existing drugs, are needed. To address this challenge, our study aimed to identify potential protein targets shared among clinically-relevant Leishmania species. We employed a subtractive proteomics and comparative genomics approach, integrating high-throughput multi-omics data to classify these targets based on different druggability metrics. This effort resulted in the ranking of 6502 ortholog groups of protein targets across 14 pathogenic Leishmania species. Among the top 20 highly ranked groups, metabolic processes known to be attractive drug targets, including the ubiquitination pathway, aminoacyl-tRNA synthetases, and purine synthesis, were rediscovered. Additionally, we unveiled novel promising targets such as the nicotinate phosphoribosyltransferase enzyme and dihydrolipoamide succinyltransferases. These groups exhibited appealing druggability features, including less than 40% sequence identity to the human host proteome, predicted essentiality, structural classification as highly druggable or druggable, and expression levels above the 50th percentile in the amastigote form. The resources presented in this work also represent a comprehensive collection of integrated data regarding trypanosomatid biology.
Graphical abstract

Highlights
Related collections
Most cited references128
- Record: found
- Abstract: found
- Article: not found
Cytoscape: a software environment for integrated models of biomolecular interaction networks.

- Record: found
- Abstract: found
- Article: found
edgeR: a Bioconductor package for differential expression analysis of digital gene expression data
- Record: found
- Abstract: found
- Article: not found