- Record: found
- Abstract: found
- Article: found
Identification of large intergenic non-coding RNAs in bovine muscle using next-generation transcriptomic sequencing
Read this article at
Abstract
Background
The advent of large-scale gene expression technologies has helped to reveal in eukaryotic cells, the existence of thousands of non-coding transcripts, whose function and significance remain mostly poorly understood. Among these non-coding transcripts, long non-coding RNAs (lncRNAs) are the least well-studied but are emerging as key regulators of diverse cellular processes. In the present study, we performed a survey in bovine Longissimus thoraci of lincRNAs (long intergenic non-coding RNAs not overlapping protein-coding transcripts). To our knowledge, this represents the first such study in bovine muscle.
Results
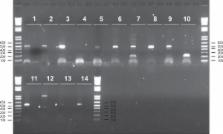
To identify lincRNAs, we used paired-end RNA sequencing (RNA-Seq) to explore the transcriptomes of Longissimus thoraci from nine Limousin bull calves. Approximately 14–45 million paired-end reads were obtained per library. A total of 30,548 different transcripts were identified. Using a computational pipeline, we defined a stringent set of 584 different lincRNAs with 418 lincRNAs found in all nine muscle samples. Bovine lincRNAs share characteristics seen in their mammalian counterparts: relatively short transcript and gene lengths, low exon number and significantly lower expression, compared to protein-encoding genes. As for the first time, our study identified lincRNAs from nine different samples from the same tissue, it is possible to analyse the inter-individual variability of the gene expression level of the identified lincRNAs. Interestingly, there was a significant difference when we compared the expression variation of the 418 lincRNAs with the 10,775 known selected protein-encoding genes found in all muscle samples. In addition, we found 2,083 pairs of lincRNA/protein-encoding genes showing a highly significant correlated expression. Fourteen lincRNAs were selected and 13 were validated by RT-PCR. Some of the lincRNAs expressed in muscle are located within quantitative trait loci for meat quality traits.
Conclusions
Our study provides a glimpse into the lincRNA content of bovine muscle and will facilitate future experimental studies to unravel the function of these molecules. It may prove useful to elucidate their effect on mechanisms underlying the genetic variability of meat quality traits. This catalog will complement the list of lincRNAs already discovered in cattle and therefore will help to better annotate the bovine genome.
Related collections
Most cited references28
- Record: found
- Abstract: found
- Article: not found
RNA maps reveal new RNA classes and a possible function for pervasive transcription.
- Record: found
- Abstract: found
- Article: not found
The evolution of gene expression levels in mammalian organs.
- Record: found
- Abstract: found
- Article: not found