- Record: found
- Abstract: found
- Article: found
SFPQ promotes an oncogenic transcriptomic state in melanoma
Read this article at
Abstract
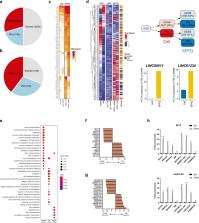
The multifunctional protein, splicing factor, proline- and glutamine-rich (SFPQ) has been implicated in numerous cancers often due to interaction with coding and non-coding RNAs, however, its role in melanoma remains unclear. We report that knockdown of SFPQ expression in melanoma cells decelerates several cancer-associated cell phenotypes, including cell growth, migration, epithelial to mesenchymal transition, apoptosis, and glycolysis. RIP-seq analysis revealed that the SFPQ-RNA interactome is reprogrammed in melanoma cells and specifically enriched with key melanoma-associated coding and long non-coding transcripts, including SOX10, AMIGO2 and LINC00511 and in most cases SFPQ is required for the efficient expression of these genes. Functional analysis of two SFPQ-enriched lncRNA, LINC00511 and LINC01234, demonstrated that these genes independently contribute to the melanoma phenotype and a more detailed analysis of LINC00511 indicated that this occurs in part via modulation of the miR-625-5p/ PKM2 axis. Importantly, analysis of a large clinical cohort revealed that elevated expression of SFPQ in primary melanoma tumours may have utility as a prognostic biomarker. Together, these data suggest that SFPQ is an important driver of melanoma, likely due to SFPQ–RNA interactions promoting the expression of numerous oncogenic transcripts.
Related collections
Most cited references70

- Record: found
- Abstract: found
- Article: found
starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data

- Record: found
- Abstract: found
- Article: found
GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis
- Record: found
- Abstract: found
- Article: not found