- Record: found
- Abstract: found
- Article: found
FunSPU: A versatile and adaptive multiple functional annotation-based association test of whole-genome sequencing data
Read this article at
Abstract
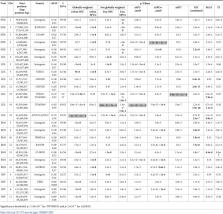
Despite ongoing large-scale population-based whole-genome sequencing (WGS) projects such as the NIH NHLBI TOPMed program and the NHGRI Genome Sequencing Program, WGS-based association analysis of complex traits remains a tremendous challenge due to the large number of rare variants, many of which are non-trait-associated neutral variants. External biological knowledge, such as functional annotations based on the ENCODE, Epigenomics Roadmap and GTEx projects, may be helpful in distinguishing causal rare variants from neutral ones; however, each functional annotation can only provide certain aspects of the biological functions. Our knowledge for selecting informative annotations a priori is limited, and incorporating non-informative annotations will introduce noise and lose power. We propose FunSPU, a versatile and adaptive test that incorporates multiple biological annotations and is adaptive at both the annotation and variant levels and thus maintains high power even in the presence of noninformative annotations. In addition to extensive simulations, we illustrate our proposed test using the TWINSUK cohort (n = 1,752) of UK10K WGS data based on six functional annotations: CADD, RegulomeDB, FunSeq, Funseq2, GERP++, and GenoSkyline. We identified genome-wide significant genetic loci on chromosome 19 near gene TOMM40 and APOC4-APOC2 associated with low-density lipoprotein (LDL), which are replicated in the UK10K ALSPAC cohort (n = 1,497). These replicated LDL-associated loci were missed by existing rare variant association tests that either ignore external biological information or rely on a single source of biological knowledge. We have implemented the proposed test in an R package “FunSPU”.
Author summary
In recent years, large-scale whole-genome sequencing (WGS) data have been generated, such as those in the UK10K project and the ongoing NIH Trans-Omics for Precision Medicine (TOPMed) WGS program, providing unprecedented opportunities to investigate low-frequency variants and rare variants in association with complex diseases and traits. However, WGS-based association analysis of complex traits remains a tremendous challenge due to the large number of rare variants, many of which are non-trait-associated neutral variants. External biological knowledge, such as functional annotations based on the ENCODE, Epigenomics Roadmap and GTEx projects, can be helpful in distinguishing causal rare variants from neutral ones; however, each functional annotation can only provide certain aspects of the biological functions. To this end, we have proposed a versatile and adaptive association test, FunSPU, to exploit multiple sources of biological knowledge in the analysis of WGS data. We illustrate our proposed test using the TWINSUK cohort of UK10K WGS data based on six functional annotations. We identified genome-wide significant genetic loci associated with low-density lipoprotein, which are replicated in the UK10K ALSPAC cohort. These replicated loci were missed by existing rare variant association tests that either ignore external biological information or rely on a single source of biological knowledge.
Related collections
Most cited references20
- Record: found
- Abstract: found
- Article: not found
Newly identified loci that influence lipid concentrations and risk of coronary artery disease.
- Record: found
- Abstract: found
- Article: not found
A SPECTRAL APPROACH INTEGRATING FUNCTIONAL GENOMIC ANNOTATIONS FOR CODING AND NONCODING VARIANTS
- Record: found
- Abstract: found
- Article: not found