- Record: found
- Abstract: found
- Article: found
Gradual Loss of ACTH Due to a Novel Mutation in LHX4: Comprehensive Mutation Screening in Japanese Patients with Congenital Hypopituitarism
Read this article at
Abstract
Mutations in transcription factors genes, which are well regulated spatially and temporally in the pituitary gland, result in congenital hypopituitarism (CH) in humans. The prevalence of CH attributable to transcription factor mutations appears to be rare and varies among populations.
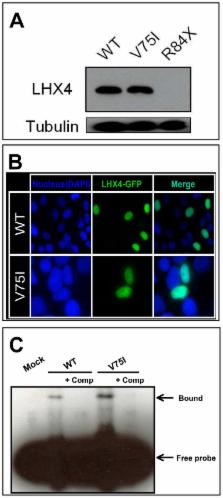
This study aimed to define the prevalence of CH in terms of nine CH-associated genes among Japanese patients. We enrolled 91 Japanese CH patients for DNA sequencing of POU1F1, PROP1, HESX1, LHX3, LHX4, SOX2, SOX3, OTX2, and GLI2. Additionally, gene copy numbers for POU1F1, PROP1, HESX1, LHX3, and LHX4 were examined by multiplex ligation-dependent probe amplification. The gene regulatory properties of mutant LHX4 proteins were characterized in vitro. We identified two novel heterozygous LHX4 mutations, namely c.249-1G>A, p.V75I, and one common POU1F1 mutation, p.R271W. The patient harboring the c.249-1G>A mutation exhibited isolated growth hormone deficiency at diagnosis and a gradual loss of ACTH, whereas the patient with the p.V75I mutation exhibited multiple pituitary hormone deficiency. In vitro experiments showed that both LHX4 mutations were associated with an impairment of the transactivation capacities of POU1F1 and αGSU, without any dominant-negative effects. The total mutation prevalence in Japanese CH patients was 3.3%. This study is the first to describe, a gradual loss of ACTH in a patient carrying an LHX4 mutation. Careful monitoring of hypothalamic–pituitary -adrenal function is recommended for CH patients with LHX4 mutations.
Related collections
Most cited references18
- Record: found
- Abstract: found
- Article: not found
Genetic screening of combined pituitary hormone deficiency: experience in 195 patients.
- Record: found
- Abstract: found
- Article: not found
A mutation in the POU-homeodomain of Pit-1 responsible for combined pituitary hormone deficiency.
- Record: found
- Abstract: found
- Article: not found