- Record: found
- Abstract: found
- Article: found
Genome sequence of the oyster mushroom Pleurotus ostreatus strain PC9
Abstract
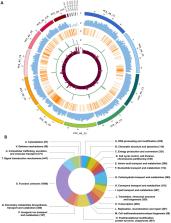
The oyster mushroom Pleurotus ostreatus is a basidiomycete commonly found in the rotten wood and it is one of the most cultivated edible mushrooms globally. Pleurotus ostreatus is also a carnivorous fungus, which can paralyze and kill nematodes within minutes. However, the molecular mechanisms of the predator–prey interactions between P. ostreatus and nematodes remain unclear. PC9 and PC15 are two model strains of P. ostreatus and the genomes of both strains have been sequenced and deposited at the Joint Genome Institute (JGI). These two monokaryotic strains exhibit dramatic differences in growth, but because PC9 grows more robustly in laboratory conditions, it has become the strain of choice for many studies. Despite the fact that PC9 is the common strain for investigation, its genome is fragmentary and incomplete relative to that of PC15. To overcome this problem, we used PacBio long reads and Illumina sequencing to assemble and polish a more integrated genome for PC9. Our PC9 genome assembly, distributed across 17 scaffolds, is highly contiguous and includes five telomere-to-telomere scaffolds, dramatically improving the genome quality. We believe that our PC9 genome resource will be useful to the fungal research community investigating various aspects of P. ostreatus biology.
Related collections
Most cited references54
- Record: found
- Abstract: found
- Article: not found
BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs.

- Record: found
- Abstract: found
- Article: found
Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement

- Record: found
- Abstract: found
- Article: found