- Record: found
- Abstract: found
- Article: found
Development of anti-PD-L1 antibody based on structure prediction of AlphaFold2
Read this article at
Abstract
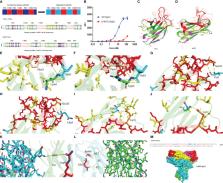
Accurate structural information plays a crucial role in comprehending biological processes and designing drugs. Indeed, the remarkable precision of the AlphaFold2 has facilitated significant advancements in predicting molecular structures, encompassing antibodies and antigens. This breakthrough has paved the way for rational drug design, ushering in new possibilities in the field of pharmaceutical development. Within this study, performing analysis and humanization guided by the structures predicted by AlphaFold2. Notably, the resulting humanized antibody, h3D5-hIgG1, demonstrated exceptional binding affinity to the PD-L1 protein. The KD value of parental antibody 3D5-hIgG1 was increased by nearly 7 times after humanization. Both h3D5-hIgG1 and 3D5-hIgG1 bound to cells expressing human PD-L1 with EC50 values of 5.13 and 9.92nM, respectively. Humanization resulted in a twofold increase in the binding capacity of the antibody, with h3D5-hIgG1 exhibiting superior performance compared to the parental antibody 3D5-hIgG1. Furthermore, h3D5-hIgG1 promoted cytokine secretion of T cells, and significantly suppressed MC38-hPD-L1 tumor growth. This study highlights the potential for artificial intelligence-assisted drug development, which is poised to become a prominent trend in the future.
Related collections
Most cited references35

- Record: found
- Abstract: found
- Article: found
Highly accurate protein structure prediction with AlphaFold
- Record: found
- Abstract: found
- Article: not found
The blockade of immune checkpoints in cancer immunotherapy.

- Record: found
- Abstract: found
- Article: found