- Record: found
- Abstract: found
- Article: found
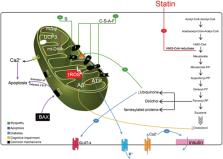
Effects of statins on mitochondrial pathways
Read this article at
Abstract
Statins are a family of drugs that are used for treating hyperlipidaemia with a recognized capacity to prevent cardiovascular disease events. They inhibit β‐hydroxy β‐methylglutaryl‐coenzyme A reductase, i.e. the rate‐limiting enzyme in mevalonate pathway, reduce endogenous cholesterol synthesis, and increase low‐density lipoprotein clearance by promoting low‐density lipoprotein receptor expression mainly in the hepatocytes. Statins have pleiotropic effects including stabilization of atherosclerotic plaques, immunomodulation, anti‐inflammatory properties, improvement of endothelial function, antioxidant, and anti‐thrombotic action. Despite all beneficial effects, statins may elicit adverse reactions such as myopathy. Studies have shown that mitochondria play an important role in statin‐induced myopathies. In this review, we aim to report the mechanisms of action of statins on mitochondrial function. Results have shown that statins have several effects on mitochondria including reduction of coenzyme Q10 level, inhibition of respiratory chain complexes, induction of mitochondrial apoptosis, dysregulation of Ca 2+ metabolism, and carnitine palmitoyltransferase‐2 expression. The use of statins has been associated with the onset of additional pathological conditions like diabetes and dementia as a result of interference with mitochondrial pathways by various mechanisms, such as reduction in mitochondrial oxidative phosphorylation, increase in oxidative stress, decrease in uncoupling protein 3 concentration, and interference in amyloid‐β metabolism.
Overall, data reported in this review suggest that statins may have major effects on mitochondrial function, and some of their adverse effects might be mediated through mitochondrial pathways.
Related collections
Most cited references172


- Record: found
- Abstract: found
- Article: found
Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel
- Record: found
- Abstract: found
- Article: not found
Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein.
Author and article information
Comments
Comment on this article
See how this article has been cited at scite.ai
scite shows how a scientific paper has been cited by providing the context of the citation, a classification describing whether it supports, mentions, or contrasts the cited claim, and a label indicating in which section the citation was made.