- Record: found
- Abstract: found
- Article: found
Recent Selective Sweeps in North American Drosophila melanogaster Show Signatures of Soft Sweeps
Read this article at
Abstract
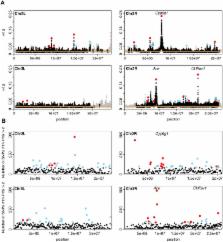
Adaptation from standing genetic variation or recurrent de novo mutation in large populations should commonly generate soft rather than hard selective sweeps. In contrast to a hard selective sweep, in which a single adaptive haplotype rises to high population frequency, in a soft selective sweep multiple adaptive haplotypes sweep through the population simultaneously, producing distinct patterns of genetic variation in the vicinity of the adaptive site. Current statistical methods were expressly designed to detect hard sweeps and most lack power to detect soft sweeps. This is particularly unfortunate for the study of adaptation in species such as Drosophila melanogaster, where all three confirmed cases of recent adaptation resulted in soft selective sweeps and where there is evidence that the effective population size relevant for recent and strong adaptation is large enough to generate soft sweeps even when adaptation requires mutation at a specific single site at a locus. Here, we develop a statistical test based on a measure of haplotype homozygosity (H12) that is capable of detecting both hard and soft sweeps with similar power. We use H12 to identify multiple genomic regions that have undergone recent and strong adaptation in a large population sample of fully sequenced Drosophila melanogaster strains from the Drosophila Genetic Reference Panel (DGRP). Visual inspection of the top 50 candidates reveals that in all cases multiple haplotypes are present at high frequencies, consistent with signatures of soft sweeps. We further develop a second haplotype homozygosity statistic (H2/H1) that, in combination with H12, is capable of differentiating hard from soft sweeps. Surprisingly, we find that the H12 and H2/H1 values for all top 50 peaks are much more easily generated by soft rather than hard sweeps. We discuss the implications of these results for the study of adaptation in Drosophila and in species with large census population sizes.
Author Summary
Evolutionary adaptation is a process in which beneficial mutations increase in frequency in response to selective pressures. If these mutations were previously rare or absent from the population, adaptation should generate a characteristic signature in the genetic diversity around the adaptive locus, known as a selective sweep. Such selective sweeps can be distinguished into hard selective sweeps, where only a single adaptive mutation rises in frequency, or soft selective sweeps, where multiple adaptive mutations at the same locus sweep through the population simultaneously. Here we design a new statistical method that can identify both hard and soft sweeps in population genomic data and apply this method to a Drosophila melanogaster population genomic dataset consisting of 145 sequenced strains collected in North Carolina. We find that selective sweeps were abundant in the recent history of this population. Interestingly, we also find that practically all of the strongest and most recent sweeps show patterns that are more consistent with soft rather than hard sweeps. We discuss the implications of these findings for the discovery and quantification of adaptation from population genomic data in Drosophila and other species with large population sizes.
Related collections
Most cited references46
- Record: found
- Abstract: not found
- Article: not found
The hitch-hiking effect of a favourable gene.
- Record: found
- Abstract: found
- Article: not found
Genomic scans for selective sweeps using SNP data.
- Record: found
- Abstract: found
- Article: not found