- Record: found
- Abstract: found
- Article: found
A Prenatally Ascertained De Novo Terminal Deletion of Chromosomal Bands 1q43q44 Associated with Multiple Congenital Abnormalities in a Female Fetus
Read this article at
Abstract
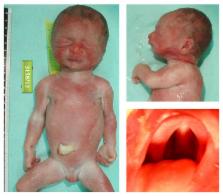
Terminal deletions in the long arm of chromosome 1 result in a postnatally recognizable disorder described as 1q43q44 deletion syndrome. The size of the deletions and the resulting phenotype varies among patients. However, some features are common among patients as the chromosomal regions included in the deletions. In the present case, ultrasonography at 22 weeks of gestation revealed choroid plexus cysts (CPCs) and a single umbilical artery (SUA) and therefore amniocentesis was performed. Chromosomal analysis revealed a possible terminal deletion in 1q and high resolution array CGH confirmed the terminal 1q43q44 deletion and estimated the size to be approximately 8 Mb. Following termination of pregnancy, performance of fetopsy allowed further clinical characterization. We report here a prenatal case with the smallest pure terminal 1q43q44 deletion, that has been molecularly and phenotypically characterized. In addition, to our knowledge this is the first prenatal case reported with 1q13q44 terminal deletion and Pierre-Robin sequence (PRS). Our findings combined with review data from the literature show the complexity of the genetic basis of the associated syndrome.
Related collections
Most cited references14
- Record: found
- Abstract: found
- Article: not found
Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence.
- Record: found
- Abstract: found
- Article: not found