- Record: found
- Abstract: found
- Article: found
GATA5 mutation homozygosity linked to a double outlet right ventricle phenotype in a Lebanese patient
Read this article at
Abstract
Background
GATA transcription factors are evolutionary conserved zinc finger proteins with multiple roles in cell differentiation/proliferation and organogenesis. GATA5 is only transiently expressed in the embryonic heart, and the inactivation of both Gata5 alleles results in a partially penetrant bicuspid aortic valve ( BAV) phenotype in mice. We hypothesized that only biallelic mutations in GATA5 could be disease causing.
Methods
A total of 185 patients with different forms of congenital heart disease ( CHD) were screened along 150 healthy individuals for GATA4, 5 , and 6. All patients' phenotypes were diagnosed with echocardiography.
Results
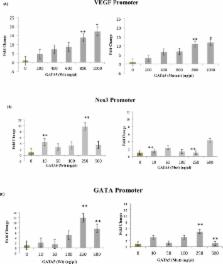
Sequencing results revealed eight missense variants (three of which are novel) in cases with various conotruncal and septal defects. Out of these, two were inherited in recessive forms: the p.T67P variant, which was found both in patients and in healthy individuals, and the previously described p.Y142H variant which was only found in a patient with a double outlet right ventricle ( DORV). We characterized the p.Y142H variant and showed that it significantly reduced the transcriptional activity of the protein over cardiac promoters by 30–40%.
Related collections
Most cited references43
- Record: found
- Abstract: found
- Article: not found
Genetics of congenital heart disease: the glass half empty.
- Record: found
- Abstract: found
- Article: not found
Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis.
- Record: found
- Abstract: found
- Article: not found