- Record: found
- Abstract: found
- Article: not found
Allosteric Modulation of the Main Protease (M Pro) of SARS-CoV-2 by Casticin—Insights from Molecular Dynamics Simulations

Read this article at

Abstract
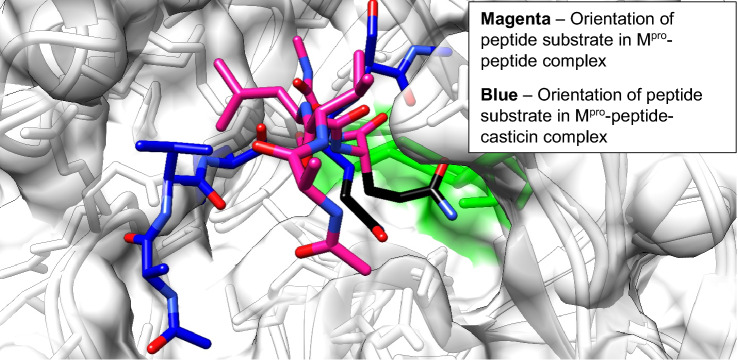
Inhibition of the main protease (M pro) of SARS-CoV-2 has been suggested to be vital in shutting down viral replication in a host. Most efforts aimed at inhibiting M Pro activity have been channeled into competitive inhibition at the active site, but this strategy will require a high inhibitor concentration and impressive inhibitor-M Pro binding affinity. Allosteric inhibition can potentially serve as an effective strategy for alleviating these limitations. In this study, the ability of antiviral natural products to inhibit M Pro in an allosteric fashion was explored with in silico techniques. Molecular docking revealed a strong interaction between casticin, an antiviral flavonoid, and M pro at a site distant from the active site. This site, characterized as a distal site, has been shown to have an interdependent dynamic effect with the active site region. M pro only, M pro-peptide (binary) and M pro-peptide-casticin (ternary) complexes were subjected to molecular dynamics simulations for 50 ns to investigate the modulatory activity of casticin binding on M pro. Molecular dynamic simulations revealed that binding of casticin at the distal site interferes with the proper orientation of the peptide substrate in the oxyanion hole of the active site, and this could lead to a halt or decrease in catalytic activity. This study therefore highlights casticin as a potential allosteric modulator of the SARS-CoV-2 main protease, which could be optimized and developed into a potential lead compound for anti-SARS-CoV-2 chemotherapy.
Related collections
Most cited references63
- Record: found
- Abstract: not found
- Article: not found
VMD: Visual molecular dynamics

- Record: found
- Abstract: found
- Article: found
SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules
- Record: found
- Abstract: found
- Article: not found