- Record: found
- Abstract: found
- Article: found
Linkage mapping and QTL analysis of growth traits in Rhopilema esculentum
Read this article at
Abstract
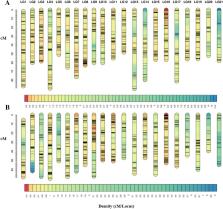
R. esculentum is a popular seafood in Asian countries and an economic marine fishery resource in China. However, the genetic linkage map and growth-related molecular markers are still lacking, hindering marker assisted selection (MAS) for genetic improvement of R. esculentum. Therefore, we firstly used 2b-restriction site-associated DNA (2b-RAD) method to sequence 152 R. esculentum specimens and obtained 9100 single nucleotide polymorphism (SNP) markers. A 1456.34 cM linkage map was constructed using 2508 SNP markers with an average interval of 0.58 cM. Then, six quantitative trait loci (QTLs) for umbrella diameter and body weight were detected by QTL analysis based on the new linkage map. The six QTLs are located on four linkage groups (LGs), LG4, LG13, LG14 and LG15, explaining 9.4% to 13.4% of the phenotypic variation. Finally, 27 candidate genes in QTLs regions of LG 14 and 15 were found associated with growth and one gene named RE13670 (sushi, von Willebrand factor type A, EGF and pentraxin domain-containing protein 1-like) may play an important role in controlling the growth of R. esculentum. This study provides valuable information for investigating the growth mechanism and MAS breeding in R. esculentum.
Related collections
Most cited references36
- Record: found
- Abstract: found
- Article: not found
KEGG: kyoto encyclopedia of genes and genomes.
- Record: found
- Abstract: found
- Article: not found
A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3.
- Record: found
- Abstract: found
- Article: not found