- Record: found
- Abstract: found
- Article: found
Systematic Review on Global Epidemiology of Methicillin-Resistant Staphylococcus pseudintermedius: Inference of Population Structure from Multilocus Sequence Typing Data
Read this article at
Abstract
Background and rationale: Methicillin-resistant Staphylococcus pseudintermedius (MRSP) is a major cause of infections in dogs, also posing a zoonotic risk to humans. This systematic review aimed to determine the global epidemiology of MRSP and provide new insights into the population structure of this important veterinary pathogen.
Methodology: Web of Science was searched systematically for articles reporting data on multilocus sequence typing (MLST) of S. pseudintermedius isolates from dogs or other animal or human patients and carriers. Data from the eligible studies were then integrated with data from the MLST database for this species. Analysis of MLST data was performed with eBURST and ClonalFrame, and the proportion of MRSP isolates resistant to selected antimicrobial drugs was determined for the most predominant clonal complexes.
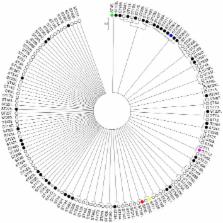
Results: Fifty-eight studies published over the last 10 years were included in the review. MRSP represented 76% of the 1428 isolates characterized by the current MLST scheme. The population of S. pseudintermedius was highly diverse and included five major MRSP clonal complexes (CCs). CC71, previously described as the epidemic European clone, is now widespread worldwide. In Europe, CC258, which is more frequently susceptible to enrofloxacin and aminoglycosides, and more frequently resistant to sulphonamides/trimethoprim than CC71, is increasingly reported in various countries. CC68, previously described as the epidemic North American clone, is frequently reported in this region but also in Europe, while CC45 (associated with chloramphenicol resistance) and CC112 are prevalent in Asia. It was estimated that clonal diversification in this species is primarily driven by homologous recombination ( r/m = 7.52).
Conclusion: This study provides evidence that S. pseudintermedius has an epidemic population structure, in which five successful MRSP lineages with specific traits regarding antimicrobial resistance, genetic diversity and geographical distribution have emerged upon a weakly clonal background through acquisition of SCC mec and other mobile genetic elements.
Related collections
Most cited references81
- Record: found
- Abstract: found
- Article: found
Arlequin (version 3.0): An integrated software package for population genetics data analysis
- Record: found
- Abstract: found
- Article: not found
eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data.
- Record: found
- Abstract: found
- Article: not found