- Record: found
- Abstract: found
- Article: found
Type V Collagen Induced Tolerance Suppresses Collagen Deposition, TGF-β and Associated Transcripts in Pulmonary Fibrosis
Read this article at
Abstract
Rationale
Idiopathic pulmonary fibrosis (IPF) is a fatal interstitial lung disease characterized by progressive scarring and matrix deposition. Recent reports highlight an autoimmune component in IPF pathogenesis. We have reported anti-col(V) immunity in IPF patients. The objective of our study was to determine the specificity of col(V) expression profile and anti-col(V) immunity relative to col(I) in clinical IPF and the efficacy of nebulized col(V) in pre-clinical IPF models.
Methods
Col(V) and col(I) expression profile was analyzed in normal human and IPF tissues. C57-BL6 mice were intratracheally instilled with bleomycin (0.025 U) followed by col(V) nebulization at pre-/post-fibrotic stage and analyzed for systemic and local responses.
Results
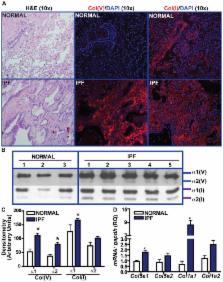
Compared to normal lungs, IPF lungs had higher protein and transcript expression of the alpha 1 chain of col(V) and col(I). Systemic anti-col(V) antibody concentrations, but not of anti-col(I), were higher in IPF patients. Nebulized col(V), but not col(I), prevented bleomycin-induced fibrosis, collagen deposition, and myofibroblast differentiation. Col(V) treatment suppressed systemic levels of anti-col(V) antibodies, IL-6 and TNF-α; and local Il-17a transcripts. Compared to controls, nebulized col(V)-induced tolerance abrogated antigen-specific proliferation in mediastinal lymphocytes and production of IL-17A, IL-6, TNF-α and IFN-γ. In a clinically relevant established fibrosis model, nebulized col(V) decreased collagen deposition. mRNA array revealed downregulation of genes specific to fibrosis ( Tgf-β, Il-1β, Pdgfb), matrix ( Acta2, Col1a2, Col3a1, Lox, Itgb1/6, Itga2/3) and members of the TGF-β superfamily ( Tgfbr1/2, Smad2/3, Ltbp1, Serpine1, Nfkb/Sp1/Cebpb).
Related collections
Most cited references32
- Record: found
- Abstract: found
- Article: not found
Pulmonary fibrosis: pathogenesis, etiology and regulation
- Record: found
- Abstract: found
- Article: not found
NADPH Oxidase-4 Mediates Myofibroblast Activation and Fibrogenic Responses to Lung Injury
- Record: found
- Abstract: found
- Article: not found