- Record: found
- Abstract: found
- Article: found
Genomic signatures underlying the oogenesis of the ectoparasitic mite Varroa destructor on its new host Apis mellifera

Read this article at
Graphical abstract
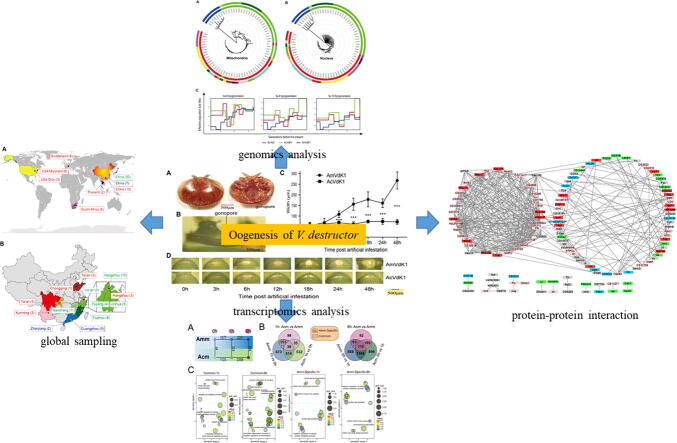
Highlights
-
•
Genes related to the oogenesis of V. destructor on their new host species were studied.
-
•
A. cerana and A. mellifera K1 mites exhibited a very close genetic relationship at the genome level.
-
•
A total of 121 genes with nonsynonymous high- F ST SNPs were found between the two types of mites.
-
•
The transcriptomes of the two types of mites differentiated as early as 1 h post-infestation.
-
•
Nine genes carrying nonsynonymous high- F ST SNPs were associated with oogenesis on the new host.
Abstract
Introduction
Host shift of parasites may have devastating effects on the novel hosts. One remarkable example is that of the ectoparasitic mite Varroa destructor, which has shifted its host from Eastern honey bees ( Apis cerana) to Western honey bees ( Apis mellifera) and posed a global threat to apiculture.
Objectives
To identify the genetic factors underlying the reproduction of host-shifted V. destructor on the new host.
Methods
Genome sequencing was conducted to construct the phylogeny of the host-shifted and non-shifted mites and to screen for genomic signatures that differentiated them. Artificial infestation experiment was conducted to compare the reproductive difference between the mites, and transcriptome sequencing was conducted to find differentially expressed genes (DEGs) during the reproduction process.
Results
The host-shifted and non-shifted V. destructor mites constituted two genetically distinct lineages, with 15,362 high- F ST SNPs identified between them. Oogenesis was upregulated in host-shifted mites on the new host A. mellifera relative to non-shifted mites. The transcriptomes of the host-shifted and non-shifted mites differed significantly as early as 1h post-infestation. The DEGs were associated with nine genes carrying nonsynonymous high- F ST SNPs, including mGluR2-like, Lamb2-like and Vitellogenin 6-like, which were also differentially expressed, and eIF4G, CG5800, Dap160 and Sas10, which were located in the center of the networks regulating the DEGs based on protein-protein interaction analysis.
Conclusions
The annotated functions of these genes were all associated with oogenesis. These genes appear to be the key genetic determinants of the oogenesis of host-shifted mites on the new host. Further study of these candidate genes will help elucidate the key mechanism underlying the success of host shifts of V. destructor.
Related collections
Most cited references50

- Record: found
- Abstract: found
- Article: found
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2
- Record: found
- Abstract: found
- Article: not found
Cytoscape: a software environment for integrated models of biomolecular interaction networks.

- Record: found
- Abstract: found
- Article: found