- Record: found
- Abstract: found
- Article: found
DNA repair factor RAD18 and DNA polymerase Polκ confer tolerance of oncogenic DNA replication stress
Read this article at
Abstract
The elevated CDK2 activity of oncogene-expressing cells induces DNA replication stress. Yang et al. show that the DNA repair protein RAD18 facilitates damage-tolerant DNA synthesis via the DNA polymerase κ in cells with aberrantly high CDK2 activity, suggesting an important new role for RAD18 in sustaining neoplastic cell survival.
Abstract
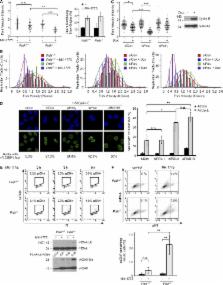
The mechanisms by which neoplastic cells tolerate oncogene-induced DNA replication stress are poorly understood. Cyclin-dependent kinase 2 (CDK2) is a major mediator of oncogenic DNA replication stress. In this study, we show that CDK2-inducing stimuli (including Cyclin E overexpression, oncogenic RAS, and WEE1 inhibition) activate the DNA repair protein RAD18. CDK2-induced RAD18 activation required initiation of DNA synthesis and was repressed by p53. RAD18 and its effector, DNA polymerase κ (Polκ), sustained ongoing DNA synthesis in cells harboring elevated CDK2 activity. RAD18-deficient cells aberrantly accumulated single-stranded DNA (ssDNA) after CDK2 activation. In RAD18-depleted cells, the G2/M checkpoint was necessary to prevent mitotic entry with persistent ssDNA. Rad18 −/− and Polκ −/− cells were highly sensitive to the WEE1 inhibitor MK-1775 (which simultaneously activates CDK2 and abrogates the G2/M checkpoint). Collectively, our results show that the RAD18–Polκ signaling axis allows tolerance of CDK2-mediated oncogenic stress and may allow neoplastic cells to breach tumorigenic barriers.
Related collections
Most cited references54
- Record: found
- Abstract: found
- Article: not found
Creation of human tumour cells with defined genetic elements.
- Record: found
- Abstract: found
- Article: not found
Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function.
- Record: found
- Abstract: found
- Article: not found