- Record: found
- Abstract: found
- Article: found
The Influence of Hepatitis C Virus Genetic Region on Phylogenetic Clustering Analysis
Read this article at
Abstract
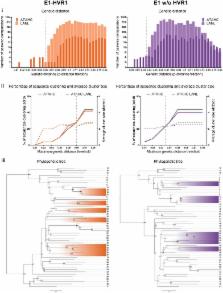
Sequencing is important for understanding the molecular epidemiology and viral evolution of hepatitis C virus (HCV) infection. To date, there is little standardisation among sequencing protocols, in-part due to the high genetic diversity that is observed within HCV. This study aimed to develop a novel, practical sequencing protocol that covered both conserved and variable regions of the viral genome and assess the influence of each subregion, sequence concatenation and unrelated reference sequences on phylogenetic clustering analysis. The Core to the hypervariable region 1 (HVR1) of envelope-2 (E2) and non-structural-5B (NS5B) regions of the HCV genome were amplified and sequenced from participants from the Australian Trial in Acute Hepatitis C (ATAHC), a prospective study of the natural history and treatment of recent HCV infection. Phylogenetic trees were constructed using a general time-reversible substitution model and sensitivity analyses were completed for every subregion. Pairwise distance, genetic distance and bootstrap support were computed to assess the impact of HCV region on clustering results as measured by the identification and percentage of participants falling within all clusters, cluster size, average patristic distance, and bootstrap value. The Robinson-Foulds metrics was also used to compare phylogenetic trees among the different HCV regions. Our results demonstrated that the genomic region of HCV analysed influenced phylogenetic tree topology and clustering results. The HCV Core region alone was not suitable for clustering analysis; NS5B concatenation, the inclusion of reference sequences and removal of HVR1 all influenced clustering outcome. The Core-E2 region, which represented the highest genetic diversity and longest sequence length in this study, provides an ideal method for clustering analysis to address a range of molecular epidemiological questions.
Related collections
Most cited references38
- Record: found
- Abstract: found
- Article: not found
Genetic diversity and evolution of hepatitis C virus--15 years on.
- Record: found
- Abstract: found
- Article: not found
Inferring species phylogenies from multiple genes: concatenated sequence tree versus consensus gene tree.
- Record: found
- Abstract: found
- Article: not found