- Record: found
- Abstract: found
- Article: found
Clinical genetics and pathobiology of ciliary chondrodysplasias
Read this article at
Abstract
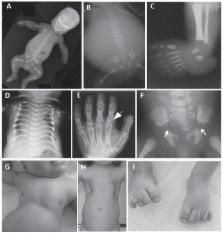
Ciliary chondrodysplasias represent a heterogenous group of rare, nearly exclusively autosomal recessively inherited developmental conditions. While the skeletal phenotype, mainly affecting limbs, ribs and sometimes the craniofacial skeleton, is predominant, extraskeletal disease affecting the kidneys, liver, heart, eyes and other organs and tissues is observed inconsistently. Significant lethality, resulting from cardiorespiratory failure due to thoracic constriction as well as from renal and hepatic insufficiency or primary cardiac failure due to congenital heart disease, is observed with these conditions. The underlying genetic defects as well as developmental biology and cell biology work undertaken using animal model systems, suggest that these rare conditions result from ciliary malfunction. The skeletal phenotype is believed to result from imbalances in the hedgehog signaling pathway that normally occurs in functional cilia in chondrocytes. Although phenotypes have been historically distinguished based on clinical features into short-rib polydactyly syndrome, Jeune asphyxiating thoracic dystrophy, Mainzer-Saldino syndrome, Sensenbrenner syndrome (cranioectodermal dysplasia), oral-facial-digital syndrome and Ellis-van Creveld syndrome, recent research suggests that there is significant genetic as well as phenotypic overlap between the conditions. This review discusses ciliary chondrodysplasias from phenotypic hallmarks to clinical management and summarizes progress in identification of the underlying molecular mechanisms as well as potential future therapeutic perspectives.
Related collections
Most cited references104
- Record: found
- Abstract: found
- Article: not found
The vertebrate primary cilium in development, homeostasis, and disease.
- Record: found
- Abstract: found
- Article: not found