- Record: found
- Abstract: found
- Article: found
Whole-genome mapping of quantitative trait loci and accuracy of genomic predictions for resistance to columnaris disease in two rainbow trout breeding populations
Read this article at
Abstract
Background
Columnaris disease (CD) is an emerging problem for the rainbow trout aquaculture industry in the US. The objectives of this study were to: (1) identify common genomic regions that explain a large proportion of the additive genetic variance for resistance to CD in two rainbow trout ( Oncorhynchus mykiss) populations; and (2) estimate the gains in prediction accuracy when genomic information is used to evaluate the genetic potential of survival to columnaris infection in each population.
Methods
Two aquaculture populations were investigated: the National Center for Cool and Cold Water Aquaculture (NCCCWA) odd-year line and the Troutlodge, Inc., May odd-year (TLUM) nucleus breeding population. Fish that survived to 21 days post-immersion challenge were recorded as resistant. Single nucleotide polymorphism (SNP) genotypes were available for 1185 and 1137 fish from NCCCWA and TLUM, respectively. SNP effects and variances were estimated using the weighted single-step genomic best linear unbiased prediction (BLUP) for genome-wide association. Genomic regions that explained more than 1% of the additive genetic variance were considered to be associated with resistance to CD. Predictive ability was calculated in a fivefold cross-validation scheme and using a linear regression method.
Results
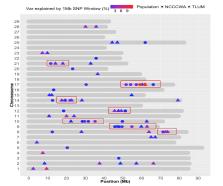
Validation on adjusted phenotypes provided a prediction accuracy close to zero, due to the binary nature of the trait. Using breeding values computed from the complete data as benchmark improved prediction accuracy of genomic models by about 40% compared to the pedigree-based BLUP. Fourteen windows located on six chromosomes were associated with resistance to CD in the NCCCWA population, of which two windows on chromosome Omy 17 jointly explained more than 10% of the additive genetic variance. Twenty-six windows located on 13 chromosomes were associated with resistance to CD in the TLUM population. Only four associated genomic regions overlapped with quantitative trait loci (QTL) between both populations.
Conclusions
Our results suggest that genome-wide selection for resistance to CD in rainbow trout has greater potential than selection for a few target genomic regions that were found to be associated to resistance to CD due to the polygenic architecture of this trait, and because the QTL associated with resistance to CD are not sufficiently informative for selection decisions across populations.
Related collections
Most cited references27
- Record: found
- Abstract: found
- Article: not found
Best linear unbiased estimation and prediction under a selection model.
- Record: found
- Abstract: found
- Article: not found
Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score.
- Record: found
- Abstract: found
- Article: not found