- Record: found
- Abstract: found
- Article: found
A Subset of Autism-Associated Genes Regulate the Structural Stability of Neurons
Read this article at
Abstract
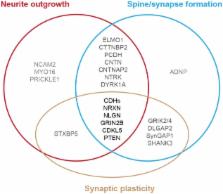
Autism spectrum disorder (ASD) comprises a range of neurological conditions that affect individuals’ ability to communicate and interact with others. People with ASD often exhibit marked qualitative difficulties in social interaction, communication, and behavior. Alterations in neurite arborization and dendritic spine morphology, including size, shape, and number, are hallmarks of almost all neurological conditions, including ASD. As experimental evidence emerges in recent years, it becomes clear that although there is broad heterogeneity of identified autism risk genes, many of them converge into similar cellular pathways, including those regulating neurite outgrowth, synapse formation and spine stability, and synaptic plasticity. These mechanisms together regulate the structural stability of neurons and are vulnerable targets in ASD. In this review, we discuss the current understanding of those autism risk genes that affect the structural connectivity of neurons. We sub-categorize them into (1) cytoskeletal regulators, e.g., motors and small RhoGTPase regulators; (2) adhesion molecules, e.g., cadherins, NCAM, and neurexin superfamily; (3) cell surface receptors, e.g., glutamatergic receptors and receptor tyrosine kinases; (4) signaling molecules, e.g., protein kinases and phosphatases; and (5) synaptic proteins, e.g., vesicle and scaffolding proteins. Although the roles of some of these genes in maintaining neuronal structural stability are well studied, how mutations contribute to the autism phenotype is still largely unknown. Investigating whether and how the neuronal structure and function are affected when these genes are mutated will provide insights toward developing effective interventions aimed at improving the lives of people with autism and their families.
Related collections
Most cited references591
- Record: found
- Abstract: found
- Article: not found
Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2.
- Record: found
- Abstract: found
- Article: not found
PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer.
- Record: found
- Abstract: found
- Article: not found