- Record: found
- Abstract: found
- Article: not found
Early-Stage Treatment with Withaferin A Reduces Levels of Misfolded Superoxide Dismutase 1 and Extends Lifespan in a Mouse Model of Amyotrophic Lateral Sclerosis
Read this article at

Abstract
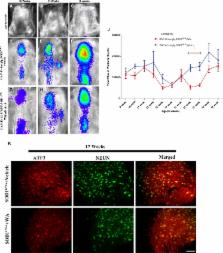
Approximately 20 % of cases of familial amyotrophic lateral sclerosis (ALS) are caused by mutations in the gene encoding Cu/Zn superoxide dismutase (SOD1). Recent studies have shown that Withaferin A (WA), an inhibitor of nuclear factor-kappa B activity, was efficient in reducing disease phenotype in a TAR DNA binding protein 43 transgenic mouse model of ALS. These findings led us to test WA in mice from 2 transgenic lines expressing different ALS-linked SOD1 mutations, SOD1 G93A and SOD1 G37R. Intraperitoneal administration of WA at a dosage of 4 mg/kg of body weight was initiated from postnatal day 40 until end stage in SOD1 G93A mice, and from 9 months until end stage in SOD1 G37R mice. The beneficial effects of WA in the SOD1 G93A mice model were accompanied by an alleviation of neuroinflammation, a decrease in levels of misfolded SOD1 species in the spinal cord, and a reduction in loss of motor neurons resulting in delayed disease progression and mortality. Interestingly, WA treatment triggered robust induction of heat shock protein 25 (a mouse ortholog of heat shock protein 27), which may explain the reduced level of misfolded SOD1 species in the spinal cord of SOD1 G93A mice and the decrease of neuronal injury responses, as revealed by real-time imaging of biophotonic SOD1 G93A mice expressing a luciferase transgene under the control of the growth-associated protein 43 promoter. These results suggest that WA may represent a potential lead compound for drug development aiming to treat ALS.
Related collections
Most cited references61
- Record: found
- Abstract: found
- Article: not found
Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation.
- Record: found
- Abstract: found
- Article: not found
A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice.
- Record: found
- Abstract: found
- Article: not found