- Record: found
- Abstract: found
- Article: found
Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer
Read this article at
SUMMARY
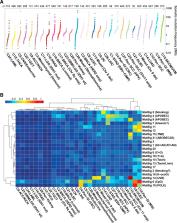
We conducted comprehensive integrative molecular analyses of the complete set of tumors in The Cancer Genome Atlas (TCGA), consisting of approximately 10,000 specimens and representing 33 types of cancer. We performed molecular clustering using data on chromosome-arm-level aneuploidy, DNA hypermethylation, mRNA, and miRNA expression levels and reverse-phase protein arrays, of which all, except for aneuploidy, revealed clustering primarily organized by histology, tissue type, or anatomic origin. The influence of cell type was evident in DNA-methylation-based clustering, even after excluding sites with known preexisting tissue-type-specific methylation. Integrative clustering further emphasized the dominant role of cell-of-origin patterns. Molecular similarities among histologically or anatomically related cancer types provide a basis for focused pan-cancer analyses, such as pan-gastrointestinal, pan-gynecological, pan-kidney, and pan-squamous cancers, and those related by stemness features, which in turn may inform strategies for future therapeutic development.
In Brief

Comprehensive, integrated molecular analysis identifies molecular relationships across a large diverse set of human cancers, suggesting future directions for exploring clinical actionability in cancer treatment.
Related collections
Most cited references20
- Record: found
- Abstract: found
- Article: found
Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma
- Record: found
- Abstract: found
- Article: not found
Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin.
- Record: found
- Abstract: found
- Article: not found