- Record: found
- Abstract: found
- Article: found
Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses

Read this article at
Summary
HIV-1-infected cells that persist despite antiretroviral therapy (ART) are frequently considered “transcriptionally silent,” but active viral gene expression may occur in some cells, challenging the concept of viral latency. Applying an assay for profiling the transcriptional activity and the chromosomal locations of individual proviruses, we describe a global genomic and epigenetic map of transcriptionally active and silent proviral species and evaluate their longitudinal evolution in persons receiving suppressive ART. Using genome-wide epigenetic reference data, we show that proviral transcriptional activity is associated with activating epigenetic chromatin features in linear proximity of integration sites and in their inter- and intrachromosomal contact regions. Transcriptionally active proviruses were actively selected against during prolonged ART; however, this pattern was violated by large clones of virally infected cells that may outcompete negative selection forces through elevated intrinsic proliferative activity. Our results suggest that transcriptionally active proviruses are dynamically evolving under selection pressure by host factors.
Graphical abstract
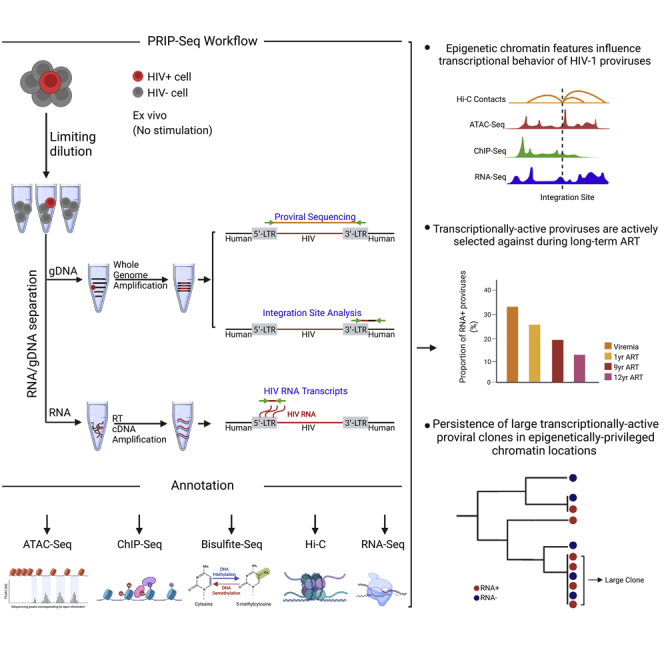
Highlights
-
•
A multidimensional assay for HIV-1 reservoir cell profiling is presented (PRIP-seq)
-
•
Transcriptionally active HIV-1 proviruses are actively selected against during ART
-
•
Large transcriptionally active proviral clones resist negative host selection forces
-
•
Epigenetic signals in linear and 3D chromatin contacts influence HIV-1 transcription
Abstract
PRIP-seq is a multidimensional single-cell assay that simultaneously captures the proviral sequence, the corresponding chromosomal integration site, and the expression of HIV-1 RNA in single virally infected cells and allows for the global mapping of transcriptionally active and silent proviruses in patients receiving suppressive antiretroviral therapy.
Related collections
Most cited references91
- Record: found
- Abstract: found
- Article: not found
Fast gapped-read alignment with Bowtie 2.

- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform
- Record: found
- Abstract: found
- Article: not found