- Record: found
- Abstract: found
- Article: found
Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy
Vanessa Schartner ,
Norma B. Romero ,
Sandra Donkervoort ,
Susan Treves ,
Pinki Munot ,
Tyler Mark Pierson ,
Ivana Dabaj ,
Edoardo Malfatti ,
Irina T. Zaharieva ,
Francesco Zorzato ,
Osorio Abath Neto ,
Guy Brochier ,
Xavière Lornage ,
Bruno Eymard ,
Ana Lía Taratuto ,
Johann Böhm ,
Hernan Gonorazky ,
Leigh Ramos-Platt ,
Lucy Feng ,
Rahul Phadke ,
Diana X. Bharucha-Goebel ,
Charlotte Jane Sumner ,
Mai Thao Bui ,
Emmanuelle Lacene ,
Maud Beuvin ,
Clémence Labasse ,
Nicolas Dondaine ,
Raphael Schneider ,
Julie Thompson ,
Anne Boland ,
Jean-François Deleuze ,
Emma Matthews ,
Aleksandra Nadaj Pakleza ,
Caroline A. Sewry ,
Valérie Biancalana ,
Susana Quijano-Roy ,
Francesco Muntoni ,
Michel Fardeau ,
Carsten G. Bönnemann ,
Jocelyn Laporte
December 23 2016
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
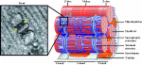
Muscle contraction upon nerve stimulation relies on excitation-contraction coupling
(ECC) to promote the rapid and generalized release of calcium within myofibers. In
skeletal muscle, ECC is performed by the direct coupling of a voltage-gated L-type
Ca(2+) channel (dihydropyridine receptor; DHPR) located on the T-tubule with a Ca(2+)
release channel (ryanodine receptor; RYR1) on the sarcoplasmic reticulum (SR) component
of the triad. Here, we characterize a novel class of congenital myopathy at the morphological,
molecular, and functional levels. We describe a cohort of 11 patients from 7 families
presenting with perinatal hypotonia, severe axial and generalized weakness. Ophthalmoplegia
is present in four patients. The analysis of muscle biopsies demonstrated a characteristic
intermyofibrillar network due to SR dilatation, internal nuclei, and areas of myofibrillar
disorganization in some samples. Exome sequencing revealed ten recessive or dominant
mutations in CACNA1S (Cav1.1), the pore-forming subunit of DHPR in skeletal muscle.
Both recessive and dominant mutations correlated with a consistent phenotype, a decrease
in protein level, and with a major impairment of Ca(2+) release induced by depolarization
in cultured myotubes. While dominant CACNA1S mutations were previously linked to malignant
hyperthermia susceptibility or hypokalemic periodic paralysis, our findings strengthen
the importance of DHPR for perinatal muscle function in human. These data also highlight
CACNA1S and ECC as therapeutic targets for the development of treatments that may
be facilitated by the previous knowledge accumulated on DHPR.
Related collections
Most cited references34
- Record: found
- Abstract: found
- Article: not found
Structure of the voltage-gated calcium channel Cav1.1 complex.
Jianping Wu, Zhen-Yu Yan, Zhangqiang Li … (2015)
- Record: found
- Abstract: found
- Article: found
T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases
Lama Al-Qusairi, Jocelyn Laporte (2011)
- Record: found
- Abstract: found
- Article: not found