- Record: found
- Abstract: found
- Article: found
Prenatally diagnosed fetal thoraco-lumbar spine duplication associated with lipomyelomeningocele: An extremely rare case of split cord malformation
case-report
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
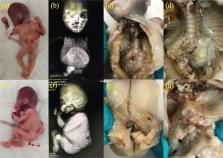
Spine duplication is considered rare, a more serious form of split cord malformation. Ultrasonographic evaluation of the spine in the second trimester is central to the antenatal diagnosis of spinal malformations. Here, we report a case of thoraco-lumbar spine duplication associated with lipomyelomeningocele diagnosed by ultrasonography at 19 weeks of gestation. To the best of our knowledge, this is the first case report of spine duplication diagnosed by antenatal ultrasonography.
Related collections
Most cited references18
- Record: found
- Abstract: found
- Article: not found
Split cord malformation: Part I: A unified theory of embryogenesis for double spinal cord malformations.
Kerith-Rae Dias, Raina Pang, M. Michael Barmada (1992)
- Record: found
- Abstract: found
- Article: not found
Split cord malformation: Part II: Clinical syndrome.
Raina Pang (1992)
- Record: found
- Abstract: found
- Article: not found
Increased DNA methylation at the AXIN1 gene in a monozygotic twin from a pair discordant for a caudal duplication anomaly.
E Whitelaw, Jack van Loon, Anita S. Chong … (2006)
Author and article information
Comments
Comment on this article
scite_