- Record: found
- Abstract: found
- Article: found
Annotated bacterial chromosomes from frame-shift-corrected long-read metagenomic data

Read this article at
Abstract
Background
Short-read sequencing technologies have long been the work-horse of microbiome analysis. Continuing technological advances are making the application of long-read sequencing to metagenomic samples increasingly feasible.
Results
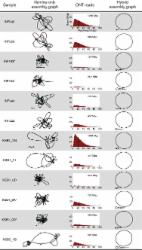
We demonstrate that whole bacterial chromosomes can be obtained from an enriched community, by application of MinION sequencing to a sample from an EBPR bioreactor, producing 6 Gb of sequence that assembles into multiple closed bacterial chromosomes. We provide a simple pipeline for processing such data, which includes a new approach to correcting erroneous frame-shifts.
Related collections
Most cited references10
- Record: found
- Abstract: found
- Article: found
Completing bacterial genome assemblies with multiplex MinION sequencing
- Record: found
- Abstract: found
- Article: not found
METAXA2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data.
Author and article information
Comments
Comment on this article
See how this article has been cited at scite.ai
scite shows how a scientific paper has been cited by providing the context of the citation, a classification describing whether it supports, mentions, or contrasts the cited claim, and a label indicating in which section the citation was made.