- Record: found
- Abstract: found
- Article: found
Genome-wide association study of 8 carcass traits in Jinghai Yellow chickens using specific-locus amplified fragment sequencing technology
Read this article at
Abstract
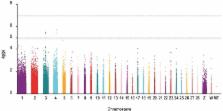
Carcass traits are important to the commercial chicken industry, and understanding the genetics of these traits will be useful in the development of commercially viable varieties of chickens. We conducted a genome-wide association study based on 8 carcass trait phenotypes in a population of 400 43-week-old Jinghai Yellow chickens. Specific-locus amplified fragment sequencing technology was used to identify 90,961 single nucleotide polymorphisms (SNP) distributed among 29 chromosomes and the mitochondrial genome. SNP that were significantly associated with phenotypic traits were identified by a simple general linear model. Fifteen SNP attained genome-wide significance ( P < 1.87E−6) and were associated with 5 of the 8 carcass traits; only one SNP was significantly associated with 2 traits (foot weight and wing weight). Twelve genes were associated with these 15 SNP. A region of chromosome 4 between 75.5 and 76.1 Mb was associated with carcass weight, foot weight, and wing weight. An 84-kb region on chromosome 3 (51.2 Mb) was associated with eviscerated weight and semi-eviscerated weight.
Related collections
Most cited references36
- Record: found
- Abstract: found
- Article: not found
Genome-wide association studies for complex traits: consensus, uncertainty and challenges.
- Record: found
- Abstract: found
- Article: not found
Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection.

- Record: found
- Abstract: found
- Article: found