- Record: found
- Abstract: found
- Article: not found
Huntingtin Interacting Proteins Are Genetic Modifiers of Neurodegeneration
Read this article at

Abstract
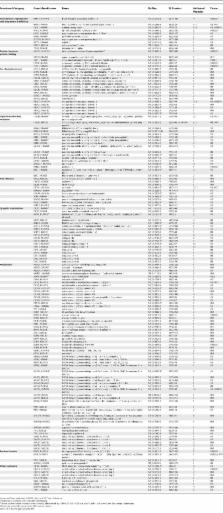
Huntington's disease (HD) is a fatal neurodegenerative condition caused by expansion of the polyglutamine tract in the huntingtin (Htt) protein. Neuronal toxicity in HD is thought to be, at least in part, a consequence of protein interactions involving mutant Htt. We therefore hypothesized that genetic modifiers of HD neurodegeneration should be enriched among Htt protein interactors. To test this idea, we identified a comprehensive set of Htt interactors using two complementary approaches: high-throughput yeast two-hybrid screening and affinity pull down followed by mass spectrometry. This effort led to the identification of 234 high-confidence Htt-associated proteins, 104 of which were found with the yeast method and 130 with the pull downs. We then tested an arbitrary set of 60 genes encoding interacting proteins for their ability to behave as genetic modifiers of neurodegeneration in a Drosophila model of HD. This high-content validation assay showed that 27 of 60 orthologs tested were high-confidence genetic modifiers, as modification was observed with more than one allele. The 45% hit rate for genetic modifiers seen among the interactors is an order of magnitude higher than the 1%–4% typically observed in unbiased genetic screens. Genetic modifiers were similarly represented among proteins discovered using yeast two-hybrid and pull-down/mass spectrometry methods, supporting the notion that these complementary technologies are equally useful in identifying biologically relevant proteins. Interacting proteins confirmed as modifiers of the neurodegeneration phenotype represent a diverse array of biological functions, including synaptic transmission, cytoskeletal organization, signal transduction, and transcription. Among the modifiers were 17 loss-of-function suppressors of neurodegeneration, which can be considered potential targets for therapeutic intervention. Finally, we show that seven interacting proteins from among 11 tested were able to co-immunoprecipitate with full-length Htt from mouse brain. These studies demonstrate that high-throughput screening for protein interactions combined with genetic validation in a model organism is a powerful approach for identifying novel candidate modifiers of polyglutamine toxicity.
Author Summary
Huntington's Disease (HD) is a fatal inherited neurodegenerative disease, which typically begins in middle age and progresses with symptoms of severe uncontrolled movements and cognitive dysfunction. HD is uniformly fatal with death occurring ten to 15 years after onset of symptoms. There is currently no effective treatment for HD. The genetic mutation underlying HD causes a protein called huntingtin (Htt) to contain an abnormally long tract of the amino acid glutamine. This extended span of glutamines changes the shape of the Htt protein, which can cause it to interact in abnormal ways with other cellular proteins. In this study, we have identified a large number of new proteins that bind to normal and mutant forms of the Htt protein. To establish a potential role for these interacting proteins in HD, we show that changing the expression of many of these proteins can modulate the pathological effects of mutant Htt on fly neurons that deteriorate when they express mutant Htt. Identifying cellular proteins that bind to Htt and modulate its pathological activity may facilitate the discovery of an effective treatment for HD.
Related collections
Most cited references57
- Record: found
- Abstract: found
- Article: not found
A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae.
- Record: found
- Abstract: found
- Article: not found
A comprehensive two-hybrid analysis to explore the yeast protein interactome.
- Record: found
- Abstract: found
- Article: not found