- Record: found
- Abstract: found
- Article: found
The performance of coalescent-based species tree estimation methods under models of missing data
Read this article at
Abstract
Background
Estimation of species trees from multiple genes is complicated by processes such as incomplete lineage sorting, gene duplication and loss, and horizontal gene transfer, that result in gene trees that differ from each other and from the species phylogeny. Methods to estimate species trees in the presence of gene tree discord due to incomplete lineage sorting have been developed and proved to be statistically consistent when gene tree discord is due only to incomplete lineage sorting and every gene tree includes the full set of species.
Results
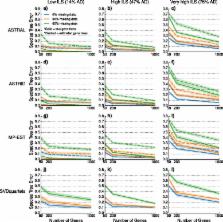
We establish statistical consistency of certain coalescent-based species tree estimation methods under some models of taxon deletion from genes. We also evaluate the impact of missing data on four species tree estimation methods (ASTRAL-II, ASTRID, MP-EST, and SVDquartets) using simulated datasets with varying levels of incomplete lineage sorting, gene tree estimation error, and degrees/patterns of missing data.
Conclusions
All the species tree estimation methods improved in accuracy as the number of genes increased and often produced highly accurate species trees even when the amount of missing data was large. These results together indicate that accurate species tree estimation is possible under a variety of conditions, even when there are substantial amounts of missing data.
Related collections
Most cited references6
- Record: found
- Abstract: not found
- Article: not found
Comparison of phylogenetic trees
- Record: found
- Abstract: found
- Article: not found
Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci.

- Record: found
- Abstract: found
- Article: found