- Record: found
- Abstract: found
- Article: found
Functional Dissection of the Enhancer Repertoire in Human Embryonic Stem Cells
Read this article at
Summary
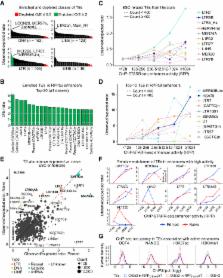
Enhancers are genetic elements that regulate spatiotemporal gene expression. Enhancer function requires transcription factor (TF) binding and correlates with histone modifications. However, the extent to which TF binding and histone modifications functionally define active enhancers remains unclear. Here, we combine chromatin immunoprecipitation with a massively parallel reporter assay (ChIP-STARR-seq) to identify functional enhancers in human embryonic stem cells (ESCs) genome-wide in a quantitative unbiased manner. Although active enhancers associate with TFs, only a minority of regions marked by NANOG, OCT4, H3K27ac, and H3K4me1 function as enhancers, with activity markedly changing under naive versus primed culture conditions. We identify an enhancer set associated with functions extending to non-ESC-specific processes. Moreover, although transposable elements associate with putative enhancers, only some exhibit activity. Similarly, within super-enhancers, large tracts are non-functional, with activity restricted to small sub-domains. This catalog of validated enhancers provides a valuable resource for further functional dissection of the regulatory genome.
Graphical Abstract

Highlights
-
•
Massively parallel reporter assay assessed over 350,000 genome regions
-
•
ChIP-STARR-seq catalogs functional enhancers in primed and naive hESCs
-
•
Identification of transcription factors and transposable elements linked to enhancers
-
•
ChIP-STARR-seq dissects super-enhancers into small functional units
Abstract
Barakat et al. use a combination of chromatin immunoprecipitation and a massively parallel reporter assay to identify functional enhancers in primed and naive human embryonic stem cells. This genome-wide catalog of validated enhancers provides a valuable resource for the further dissection of the regulatory genome.
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
A unique chromatin signature uncovers early developmental enhancers in humans.
- Record: found
- Abstract: found
- Article: not found
Transcription factors: from enhancer binding to developmental control.

- Record: found
- Abstract: found
- Article: found