- Record: found
- Abstract: found
- Article: found
Degradation of edible mushroom waste by Hermetia illucens L. and consequent adaptation of its gut microbiota
Read this article at
Abstract
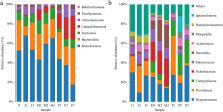
The edible fungus industry is one of the pillar industries in the Yunnan–Guizhou Plateau, China. The expansion of the planting scale has led to the release of various mushroom residues, such as mushroom feet, and other wastes, which are not treated adequately, resulting in environmental pollution. This study investigated the ability of black soldier fly ( Hermetia illucens L.) larvae (BSFL) to degrade mushroom waste. Moreover, this study analyzed changes in the intestinal bacterial community and gene expression of BSFL after feeding on mushroom waste. Under identical feeding conditions, the remaining amount of mushroom waste in Pleurotus ostreatus treatment group was reduced by 18.66%, whereas that in Flammulina velutipes treatment group was increased by 31.08%. Regarding gut microbial diversity, compared with wheat bran-treated control group, Dysgonomonas, Providencia, Enterococcus, Pseudochrobactrum, Actinomyces, Morganella, Ochrobactrum, Raoultella, and Ignatzschineria were the most abundant bacteria in the midgut of BSFL in F. velutipes treatment group. Furthermore, Dysgonomonas, Campylobacter, Providencia, Ignatzschineria, Actinomyces, Enterococcus, Morganella, Raoultella, and Pseudochrobactrum were the most abundant bacteria in the midgut of BSFL in P. ostreatus treatment group. Compared with wheat bran-treated control group, 501 upregulated and 285 downregulated genes were identified in F. velutipes treatment group, whereas 211 upregulated and 43 downregulated genes were identified in P. ostreatus treatment group. Using Kyoto Encyclopedia of Genes and Genomes and Gene Ontology enrichment analyses, we identified 14 differentially expressed genes (DEGs) related to amino sugar and nucleotide sugar metabolism in F. velutipes treatment group, followed by 12 DEGs related to protein digestion and absorption. Moreover, in P. ostreatus treatment group, two DEGs were detected for fructose and mannose metabolism, and two were noted for fatty acid metabolism. These results indicate that feeding on edible mushroom waste can alter the intestinal microbial community structure of BSFL; moreover, the larval intestine can generate a corresponding feedback. These changes contribute to the degradation of edible mushroom waste by BSFL and provide a reference for treating edible mushroom waste using BSFL.
Related collections
Most cited references45

- Record: found
- Abstract: found
- Article: found
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2
- Record: found
- Abstract: found
- Article: not found
Gene Ontology: tool for the unification of biology
- Record: found
- Abstract: found
- Article: not found