- Record: found
- Abstract: found
- Article: found
Specific Loss of Histone H3 Lysine 9 Trimethylation and HP1γ/Cohesin Binding at D4Z4 Repeats Is Associated with Facioscapulohumeral Dystrophy (FSHD)
Read this article at
Abstract
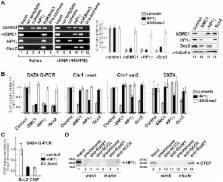
Facioscapulohumeral dystrophy (FSHD) is an autosomal dominant muscular dystrophy in which no mutation of pathogenic gene(s) has been identified. Instead, the disease is, in most cases, genetically linked to a contraction in the number of 3.3 kb D4Z4 repeats on chromosome 4q. How contraction of the 4qter D4Z4 repeats causes muscular dystrophy is not understood. In addition, a smaller group of FSHD cases are not associated with D4Z4 repeat contraction (termed “phenotypic” FSHD), and their etiology remains undefined. We carried out chromatin immunoprecipitation analysis using D4Z4–specific PCR primers to examine the D4Z4 chromatin structure in normal and patient cells as well as in small interfering RNA (siRNA)–treated cells. We found that SUV39H1–mediated H3K9 trimethylation at D4Z4 seen in normal cells is lost in FSHD. Furthermore, the loss of this histone modification occurs not only at the contracted 4q D4Z4 allele, but also at the genetically intact D4Z4 alleles on both chromosomes 4q and 10q, providing the first evidence that the genetic change (contraction) of one 4qD4Z4 allele spreads its effect to other genomic regions. Importantly, this epigenetic change was also observed in the phenotypic FSHD cases with no D4Z4 contraction, but not in other types of muscular dystrophies tested. We found that HP1γ and cohesin are co-recruited to D4Z4 in an H3K9me3–dependent and cell type–specific manner, which is disrupted in FSHD. The results indicate that cohesin plays an active role in HP1 recruitment and is involved in cell type–specific D4Z4 chromatin regulation. Taken together, we identified the loss of both histone H3K9 trimethylation and HP1γ/cohesin binding at D4Z4 to be a faithful marker for the FSHD phenotype. Based on these results, we propose a new model in which the epigenetic change initiated at 4q D4Z4 spreads its effect to other genomic regions, which compromises muscle-specific gene regulation leading to FSHD pathogenesis.
Author Summary
Most cases of facioscapulohumeral muscular dystrophy (FSHD) are associated with a decrease in the number of D4Z4 repeat sequences on chromosome 4q. How this leads to the disease remains unclear. Furthermore, D4Z4 shortening is not seen in a small number of FSHD cases, and the etiology is unknown. In the cell, the DNA, which encodes genetic information, is wrapped around abundant nuclear proteins called histones to form a “beads on a string”–like structure termed chromatin. It became apparent that these histones are modified to regulate both maintenance and expression of genetic information. In the current study, we characterized the chromatin structure of the D4Z4 region in normal and FSHD patient cells. We discovered that one particular histone modification (trimethylation of histone H3 at lysine 9) in the D4Z4 repeat region is specifically lost in FSHD. We identified the enzyme responsible for this modification and the specific factors whose binding to D4Z4 is dependent on this modification. Importantly, these chromatin changes were observed in both types of FSHD, but not in other muscular dystrophies. Thus, this chromatin abnormality at D4Z4 unifies the two types of FSHD, which not only serves as a novel diagnostic marker, but also provides new insight into the role of chromatin in FSHD pathogenesis.
Related collections
Most cited references53
- Record: found
- Abstract: found
- Article: not found
Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain.
- Record: found
- Abstract: found
- Article: not found
Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells.
- Record: found
- Abstract: found
- Article: not found