- Record: found
- Abstract: found
- Article: found
Identifying plant genes shaping microbiota composition in the barley rhizosphere
Read this article at
Abstract
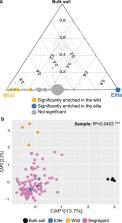
A prerequisite to exploiting soil microbes for sustainable crop production is the identification of the plant genes shaping microbiota composition in the rhizosphere, the interface between roots and soil. Here, we use metagenomics information as an external quantitative phenotype to map the host genetic determinants of the rhizosphere microbiota in wild and domesticated genotypes of barley, the fourth most cultivated cereal globally. We identify a small number of loci with a major effect on the composition of rhizosphere communities. One of those, designated the QRMC-3HS, emerges as a major determinant of microbiota composition. We subject soil-grown sibling lines harbouring contrasting alleles at QRMC-3HS and hosting contrasting microbiotas to comparative root RNA-seq profiling. This allows us to identify three primary candidate genes, including a Nucleotide-Binding-Leucine-Rich-Repeat ( NLR) gene in a region of structural variation of the barley genome. Our results provide insights into the footprint of crop improvement on the plant’s capacity of shaping rhizosphere microbes.
Abstract
A prerequisite to exploiting soil microbes for sustainable crop production is the identification of the plant genes shaping microbiota composition in the rhizosphere. Here, the authors report QTLs and the associated candidate genes underlying rhizosphere microbiome composition in barley.
Related collections
Most cited references105

- Record: found
- Abstract: found
- Article: found
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2
- Record: found
- Abstract: found
- Article: not found
STAR: ultrafast universal RNA-seq aligner.
- Record: found
- Abstract: found
- Article: not found