- Record: found
- Abstract: found
- Article: found
Population‐based genetic testing for cancer susceptibility genes: quo vadis?
article-commentary
08 September 2022
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Ovarian cancer (OC), breast cancer (BC), endometrial cancer (EC) and colorectal cancer
(CRC) account for approximately 50% of cancers in women.
1
A total of 2.9 million women worldwide and approximately 88 000 women in the UK are
diagnosed with these cancers annually, and 1.05 million women worldwide and 25 000
women in the UK die from them per year.
1
,
2
GLOBOCAN predicts that the number of these cancer cases will rise by 27%–53% worldwide
(and by 20%–36% in women in the UK) and deaths by 49%–69% worldwide (and by 36%‐47%
in women in the UK) over the next 20 years.
2
‘Pathogenic and likely pathogenic variants’, herein termed ‘pathogenic variants’ or
‘PVs’, in a number of high–moderate penetrance cancer susceptibility genes (CSGs)
can cause high‐risk breast and/or ovarian cancer syndrome or Lynch syndrome (caused
by mismatched repair genes). High‐risk breast and ovarian cancer syndrome is associated
with an increased risk of developing BC and/or OC. Lynch syndrome is associated mainly
with an increased risk of CRC, EC and OC (see Table 1). Overall, CSGs account for
around 15%–20% of OC,
3
4% of BC,
4
3% of EC,
5
and 3%–4% of CRC,
6
,
7
and a majority of these cancers are potentially preventable. High‐risk breast and
ovarian cancer syndrome and Lynch syndrome fall under tier‐1 genomic applications,
defined by the Centers for Disease Control and Prevention and the Office of Public
Health Genomics as those having significant potential for positive impact on public
health based on existing evidence‐based guidelines and recommendations. Effective
preventive therapy options, including risk‐reducing surgery (mastectomy, risk‐reducing
salpingo‐oophorectomy (RRSO) or hysterectomy), chemoprevention (e.g. aspirin or selective
estrogen receptor modulators) and screening (for women at high risk of BC or CRC),
to reduce these CSG carrier‐associated cancer risks are available in the UK National
Health Service (NHS) and other health systems (Table 1). Women can also make lifestyle,
contraceptive and reproductive choices, including prenatal/pre‐implantation genetic
diagnosis, all of which can impact cancer risk.
TABLE 1
Tier‐1 syndromes, cancer susceptibility genes, cancer risks and management options
Genes
Cancer risks %
Risk‐management options
BC
OC
CRC
EC
BC
OC
CRC
EC
Other
HBOC
BRCA1
72
44
RRM, chemoprevention (SERM, aromatase inhibitors), screening (MRI, mammogram)
a
RRSO, RRESDO
Lifestyle, reproduction, contraception, PND, PGD
BRCA2
69
17
PALB2
53
5
RAD51C
21
11
Screening (mammogram)
a
RAD51D
20
13
BRIP1
6
LS
MLH1
11
48
37
Hysterectomy & BSO
Screening (colonoscopy), chemoprevention (aspirin), surgical prevention
Hysterectomy, annual USS, hysteroscopy & endometrial biopsy
MSH2
17
47
49
MSH6
11
20
41
PMS2
b
3
10
13
Abbreviations: BSO, bilateral salpingo‐oophorectomy; CP, chemoprevention; HBOC, high‐risk
breast and/or ovarian cancer syndrome; LS, Lynch syndrome; PGD, pre‐implantation genetic
diagnosis; PND, prenatal diagnosis; RRESDO, risk‐reducing early salpingectomy and
delayed oophorectomy; RRM, risk‐reducing mastectomy; RRSO, risk‐reducing salpingo‐oophrectomy;
SERM, selective estrogen receptor modulators.
a
NHS High Risk Breast Cancer Screening Programme.
b
BSO is not recommended for PMS2 as ovarian cancer risk is similar to population‐level
risk.
The traditional model of genetic testing to identify CSG carriers involves accessing
genetic testing through high‐risk cancer genetics clinics/services and is based on
fulfilling a strong three‐generational family history or standardized clinical criteria.
This process is complex, can vary regionally and internationally, and has been shown
to be hampered by limited public and health professional awareness, restricted access,
inadequate uptake and a huge underutilisation of genetic testing. Besides family history,
clinical criteria are only moderately effective at identifying PV carriers and have
an extremely poor ability to rule out a PV carrier.
8
Additionally, the traditional genetic testing thresholds have been set too high (e.g.
10% combined probability for ‘BRCA1 and BRCA2’ testing). We and others have shown
that around 50% of breast and ovarian CSG carriers do not fulfil current clinical/family
history‐based genetic testing criteria, and are missed.
3
,
9
,
10
Far greater numbers of carriers are missed through population‐based ascertainment.
11
For Lynch syndrome, the Bethesda molecular criteria and Amsterdam‐II clinical criteria
miss 12%–30% and 55%–70% of carriers, respectively.
5
Recent data show that traditional family history guidelines may further magnify health
inequalities for minority communities like non‐Hispanic Black populations, by identifying
proportionally fewer high‐risk women in these populations.
12
We showed that despite 25 years of a well‐structured national service for clinical
genetics, free at the point of care, over 97% of BRCA carriers remain undetected in
a population of 16 million in London.
13
Forecasting models suggest that current detection rates are inadequate, and even doubling
the rates would take 165 years to identify the ‘clinically detectable’ proportion
of BRCA carriers, with 50% remaining unidentifiable as they don't fulfil the testing
criteria. Given the effective risk management including screening (for BC/CRC) and
the preventive therapy options available for CSG carriers, this highlights the inadequacy
of our current approach and the massive scale of missed opportunities for cancer prevention.
Next‐generation sequencing technologies, falling costs, advancements in bioinformatics,
our increasing understanding and applicability of genetics, coupled with rising public
awareness, now permits large‐scale, high‐throughput, population‐based genetic testing
(‘population testing’). Why should we wait for someone to develop cancer in order
to identify people in whom we can prevent cancer? Identifying a woman as a CSG carrier
after she develops cancer is a failure of cancer prevention!
Changing the paradigm to population testing can address the limitations in the current
clinical genetic testing model for CSGs across health systems and provides a forward‐looking
strategy to maximise precision prevention. Precision prevention encompasses a prevention
strategy that incorporates individual variation in genetic, epigenetic and non‐genetic
(environmental, hormonal, reproductive and lifestyle) factors. Half a century ago
Wilson and Jungner provided the initial guiding principles for population testing
for disease.
14
These have been modified over the years and the UK National Screening Committee has
established criteria for UK screening programmes. Over the years, additional adaptations
to these principles have been developed for screening for genetic susceptibility,
including important principles such as ‘analytic validity, clinical validity, clinical
utility and associated ethical, legal and social implications’ (ACCE framework),
15
and other modifications.
The development of any population‐testing framework needs to consider both benefits
and harms and only include testing for CSGs with well‐established clinical utility.
There should be effective interventions to reduce cancer risk and the risk conferred
by the CSGs should lie above the risk thresholds for undertaking these interventions.
For example, RRSO is now recommended for women at greater than 4%–5% lifetime OC risk
in the UK,
16
or at greater than 3%–4% lifetime OC risk in the USA,
17
thus providing clinical utility for testing newer, moderate‐penetrance CSGs.
1
THE JEWISH MODEL FOR POPULATION‐BASED GENETIC TESTING (POPULATION TESTING)
The greatest wealth of data supporting population testing comes from BRCA‐testing
in the Jewish population. Around 1 in 40 Ashkenazi Jewish (AJ) individuals carry one
of three Jewish BRCA founder mutations.
9
,
18
Our UK randomised trial (GCaPPS) showed that population‐based BRCA‐testing (compared
with family history‐based testing) in the AJ community is feasible, acceptable, safe,
has high satisfaction, does not harm quality of life or psychological well‐being,
reduces long‐term anxiety, reduces uncertainty, more than doubles the BRCA carriers
identified, and can be delivered in a community setting.
9
,
19
These findings are corroborated and complemented by data from large‐cohort studies
from Australia, Canada, Israel and the USA.
18
,
20
Jewish population BRCA‐testing has been demonstrated to be extremely cost‐effective
and in fact is cost saving in most scenarios.
21
In all, 10% of BC and 40% of OC in the Jewish population are caused by BRCA founder
mutations and are potentially preventable.
22
,
23
We and others have long advocated changing policy to offer population‐based BRCA‐testing
in the Jewish community. Consequently, Israel has recently changed policy and now
offers population BRCA founder mutation testing to all Jewish individuals. Pilot sites
offering BRCA‐testing for the Jewish population are expected to be implemented in the
UK health service in 2023. The Jewish population is the first population worldwide
to undergo population testing in a clinical/healthcare setting.
2
BIOBANKS/GENOMIC POPULATION COHORTS
Additional secondary findings, including PVs in CSGs, have been returned to patients/populations
recruited to large biobanks and/or population cohorts, for example the UK Biobank,
the 100,000 Genomes Project, the Geisenger MyCode Initiative, the LifePool Study and
the Healthy Nevada Project. Although these data are complementary, add to the increasing
evidence base and address the population PV prevalence for established CSGs, this
bolt‐on return of undertaking additional ‘secondary findings’ is not equivalent to
the prospective uptake of testing CSGs in an unselected unaffected population. A selective
subgroup opting for the return of incidental/secondarily looked for findings is not
generalisable to an unselected unaffected general population. Post‐hoc sequencing
and/or analysis does not address, in a prospective unbiased fashion, the key issues
and problems related to: (i) the logistics of population testing; (ii) information
giving, consent and the uptake of testing; (iii) the uptake of screening and preventive
options; (iv) the management of variants of unknown significance; and (v) the long‐term
outcomes.
3
POPULATION TESTING IN THE GENERAL POPULATION
Findings from the AJ population cannot be directly extrapolated to the general non‐Jewish
population. The Canadian ‘Screen Project’ provided a direct‐to‐consumer BRCA‐testing
option in the general population and has been the first of its kind. However, participants
(rather than the health system) were expected to pay for their test through out‐of‐pocket
costs. A total of 1269 individuals were tested over 2 years. Although this approach
may be helpful for improving access for some, a health system‐funded population screening
programme is what is needed to maximise uptake, to ensure equity of access and downstream
management, and to maximise the population impact. We demonstrated the potential cost‐effectiveness
and beneficial population impact of population BRCA‐testing across multiple health
systems in high‐income and upper/middle‐income countries.
24
This approach is potentially cost‐saving for the Netherlands and the USA, and is cost‐effective
for the UK, Brazil and China.
24
The cost of testing needs to fall further for it to be cost‐effective in low‐income
countries like India.
24
This strategy can prevent tens of thousands more BC and OC cases compared with current
clinical strategies. We estimate the total general population prevalence of tier‐1
CSGs associated with BRCA1, BRCA2, PALB2, RAD51C, RAD51D, BRIP1, MLH1, MSH2, MSH6
and PMS2 CSGs, listed in Table 1, to be around 1.3%.
25
,
26
Data from large biobank/cohort studies show that approximately 75% of CSG carriers
do not fulfil traditional family history‐based clinical criteria and would be missed.
11
Relatives of PV carriers identified can undergo cascade testing. Unaffected relatives
of PV carriers identified through cascade testing can also access risk management
and preventive interventions (Table 1). Not all CSG carriers identified will develop
cancer as these genes have variable penetrance. All at‐risk individuals should have
informed counselling of the pros and cons of risk‐management options, including surgical
prevention. Undergoing preventive surgery can be a complex and difficult decision‐making
process, which changes with time. Different individuals may opt for it at different
time points, and some may make an informed choice not to undergo it. Expanding on
our earlier modelling with current clinical uptake rates for surgical prevention,
we estimate that testing 10 000 women could potentially lead to preventing, in total,
approximately 210 cases of BC, OC, EC and CRC combined.
24
,
25
We previously demonstrated the cost‐effectiveness of population‐based testing for
a panel of tier‐1 high‐risk breast/ovarian CSGs genes (BRCA1, BRCA2, RAD51C, RAD51D,
BRIP1 and PALB2) in the UK and USA healthcare settings, with an incremental cost‐effectiveness
ratio (ICER) of £21,599.96 per quality‐adjusted life‐year (QALY), or $54,769.78/QALY,
with 83.7% and 92.7% of simulations being cost‐effective on probabilistic sensitivity
analysis.
25
The potential cost‐effectiveness of testing for BRCA1, BRCA2, MLH1, MSH2, FXS and
CF has also been highlighted for the Australian population.
27
Complex risk models incorporating genetic, family history, epidemiological and clinical
variables are now being used to predict personalised absolute cancer risk. These have
been developed and validated for a number of cancers, including BC, EC and OC. Although
good validation data are available for BC and are beginning to emerge for OC, more
robust validation data are needed for other cancers. This approach enables population
stratification for risk‐adapted screening and/or risk‐adapted prevention. BC risk
models incorporating a single‐nucleotide polymorphism (SNP)‐based polygenic risk score
(PRS), mammographic density and epidemiological variables are currently being used
to implement risk‐adapted BC screening in large‐scale population cohorts (UK PROCAS
study) and in clinical trials such as WISDOM (USA) and MyPeBS (European). Our pilot
population‐testing study to predict personalised OC risk using a validated OC‐risk
model incorporating CSGs, PRS and epidemiological/reproductive risk factors recruited
women through primary care using a web‐based decision tool, and demonstrated feasibility,
acceptability, high satisfaction and a reduction in cancer worry with this approach.
28
More real‐world multidisciplinary implementation studies are needed to evaluate the
impact of population testing for CSGs. Research needs to evaluate the psychological
and socio‐ethical outcomes of population testing. Although initial modelling has highlighted
the potential cost‐effectiveness of this approach, real‐world studies with long‐term
outcomes of screening and prevention are needed to confirm that the model assumptions
are valid and will translate to patient benefit and a reduction in cancer incidence,
reconfirming the cost‐effectiveness. It is likely that population‐testing implementation
models will vary by country and health system, as they will need to be context specific
while following the common core principles of population testing (for an example,
see Figure 1). The simplification and mainstreaming of such large‐scale testing will
require the digitisation of the process of information giving, consent and a direct‐to‐patient
(saliva‐based) testing approach, with more intensive counselling and support reserved
for those testing positive.
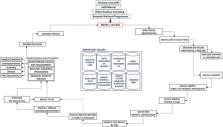
FIGURE 1
Population‐based testing pathway
Other challenges that need to be tackled include a method for the management of variants
of unknown significance and developing a structure or framework for safe data management,
data protection, consenting and the delivery of results. Subsequent scaling up for
implementation across the health system will have additional challenges, including
stakeholder engagement, awareness campaigns, an expansion in health workforce infrastructure,
laboratory/testing services, and downstream screening and prevention infrastructure.
The future potential for population testing to maximise precision prevention globally
across high‐income, middle‐income and low‐income health systems is exciting and bright.
The costs of genetic testing have fallen tenfold over the last decade. Although currently
cost‐effective for high/middle‐income countries, a price point of approximately $100
a test can make this approach potentially affordable in low‐income countries too.
We believe that this will be achievable in the future.
Two prospective general population‐testing studies are being implemented over the
next year that will provide an initial evidence base for the assessment of population
testing. The Australian ‘DNA screen pilot study’ will recruit 10 000 healthy individuals
between 18 and 40 years of age through social media and offer testing for high‐risk
BC/OC, Lynch syndrome and familial hypercholesterolaemia CSGs.
29
Our UK PROTECT (population‐based germline testing for early detection and cancer prevention)
trial will evaluate the impact of implementing a population‐based panel genetic testing
strategy for high‐ and moderate‐penetrance high‐risk BC/OC and Lynch syndrome CSGs
in more than 5000 women aged >18 years recruited through primary care using a web‐based
digitally enabled direct‐to‐patient saliva‐based DNA testing approach. PROTECT will
address current knowledge gaps for population testing by evaluating the incremental
PVs detected, uptake of testing, acceptability, satisfaction, psychosocial well‐being,
overall impact, socio‐ethics, management strategy for variants of unknown significance,
long‐term uptake of screening and prevention interventions, and health‐economic outcomes
of population‐based genetic testing.
ACKNOWLEDGEMENT
None.
AUTHOR CONTRIBUTIONs
RM and MS drafted and wrote the commentary.
FUNDING INFORMATION
This work is supported by a grant from Yorkshire Cancer Research.
CONFLICT OF INTERESTS
RM is currently funded by Yorkshire Cancer Research for research into population testing.
RM has previously received research funding from The Eve Appeal and Cancer Research
UK into population testing. RM is supported by an NHS Innovation Accelerator (NIA)
Fellowship for population testing. RM has research funding from Barts & the London
Charity, Rose Trees Trust and BGCS outside this work, an honorarium for grant review
from Israel National Institute for Health Policy Research and an honorarium for advisory
board membership or lectures from Astrazeneca/MSD/GSK/EGL. Completed disclosure of
interests form available to view online as supporting information.
Supporting information
Appendix S1
Click here for additional data file.
Appendix S2
Click here for additional data file.
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
Feasibility of screening for Lynch syndrome among patients with colorectal cancer.
Heather Hampel, Wendy Frankel, Edward Martin … (2008)
- Record: found
- Abstract: found
- Article: not found
Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2.
Mary-Claire King, Joan Marks, Jessica Mandell (2003)
- Record: found
- Abstract: found
- Article: not found
Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer
Robert MacInnis, Noralane Lindor, Polly Newcomb … (2017)