- Record: found
- Abstract: found
- Article: found
A DNA barcode-based survey of wild urban bees in the Loire Valley, France
Read this article at
Abstract
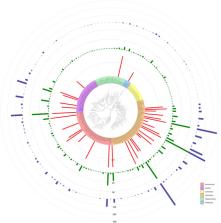
The current decline of wild bees puts important ecosystem services such as pollination at risk. Both inventory and monitoring programs are needed to understand the causes of wild bee decline. Effective insect monitoring relies on both mass-trapping methods coupled with rapid and accurate identifications. Identifying wild bees using only morphology can be challenging, in particular, specimens from mass-trapped samples which are often in poor condition. We generated DNA barcodes for 2931 specimens representing 157 species (156 named and one unnamed species) and 28 genera. Automated cluster delineation reveals 172 BINs (Barcodes Index Numbers). A total of 36 species (22.93%) were found in highly urbanized areas. The majority of specimens, representing 96.17% of the species barcoded form reciprocally exclusive groups, allowing their unambiguous identification. This includes several closely related species notoriously difficult to identify. A total of 137 species (87.26%) show a “one-to-one” match between a named species and the BIN assignment. Fourteen species (8.92%) show deep conspecific lineages with no apparent morphological differentiation. Only two species pairs shared the same BIN making their identification with DNA barcodes alone uncertain. Therefore, our DNA barcoding reference library allows reliable identification by non-experts for the vast majority of wild bee species in the Loire Valley.
Related collections
Most cited references73

- Record: found
- Abstract: found
- Article: found
FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments
- Record: found
- Abstract: found
- Article: not found
MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform.

- Record: found
- Abstract: found
- Article: found