- Record: found
- Abstract: not found
- Article: not found
A 336-Nucleotide In-Frame Deletion in ORF7a Gene of SARS-CoV-2 Identified in Genomic Surveillance by Next-Generation Sequencing
letter
Read this article at

There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Public health laboratories (PHLs) across the US have been taking a central role in
the response to the coronavirus disease 2019 (COVID-19) pandemic [1]. Currently, PHLs
have increased their capacity to sequence whole genomes of the severe acute respiratory
syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19, which enhances
surveillance. SARS-CoV-2 genomes are known to use non-canonical translation mechanisms
such as leaky scanning, ribosomal frameshifting, and alternative initiation, as is
commonly observed in the coronavirus family 2, 3, 4, 5, 6. Here, we report the longest
in-frame deletion in ORF7a gene identified in a patient using Next-Generation Sequencing
(NGS) during baseline surveillance.
A nasal sample was collected on July 7th, 2021, from a previously tested SARS-CoV-2
positive pediatric asymptomatic female in San Mateo County Public Health Laboratory,
California, USA. The presence of SARS-CoV-2 in a specimen was confirmed by the CDC
2019-Novel Coronavirus Real-Time RT-PCR Diagnostic Panel, that targets two unique
regions in the SARS-CoV-2 nucleocapsid gene (N1 and N2) and the human RNase P gene
(RP) [7], with the cycle threshold values of 15.4, 15.6, and 26.8, respectively. Subsequently,
the total nucleic acids from the SARS-CoV-2 positive specimen were used for NGS library
preparation utilizing the Respiratory Pathogen ID/AMR Enrichment Panel (RPIP) Kit
(Illumina). The concentration and distribution size of the prepared library was checked
on a Qubit 4 fluorometer (Life Technologies) and 4200 TapeStation (Agilent Technologies),
respectively. Paired-end NGS was performed on an Illumina MiniSeq instrument using
the 150-cycle MiniSeq high output reagent kit (Illumina). Sequence data were assembled
using the Explify RPIP Analysis App v.3.2.8 (IDbyDNA) and validated by the viral-ngs
v.2.1.8 workflows (Broad Viral Genomics) on the Terra platform (app.terra.bio) [8].
Briefly, the SARS-CoV-2 genome was assembled using a reference-based assembly approach
of quality-filtered raw reads with the reference genome Wuhan-Hu-1 (NC_045512.2) [9].
A total of 8,363,544 raw paired-end reads were generated. The genome consensus of
the isolate, designated CA-SMCPHL-072321.3, was called from 2,071,958 SARS-CoV-2 reads
using the minimap2 aligner [10]. This resulted in the mean read depth coverage of
5,066 reads. The final genome consensus was 29,548 nucleotides (nt) long and was deposited
in GISAID under the accession number EPI_ISL_6159211. The genome was classified as
Nextstrain clade 20I [11] from Pango lineage B.1.1.7 [12]. During the annotation of
this genome, we noticed a 336-nt deletion in the gene-encoding accessory protein ORF7a
(27,418-27,753) (Figure 1
). Although numerous studies have been performed for some other SARS-CoV-2 viral proteins
13, 14, 15, 16, 17, studies on putative activity and role of ORF7a is just starting
to arise. It has been demonstrated that SARS-CoV-2 ORF7a protein inhibits type I interferon
(IFN-1) signaling [18, 19], interacts with CD14+ monocytes [20], induces the nuclear
factor kappa B (NF-κB) pathway [21], and thus triggers expression of proinflammatory
cytokines, including interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α).
This forms the basis of a likely mechanism through which ORF7a mediates the potentially
fatal cytokine storm progression, indicating that ORF7a may be a key viral factor
for clinical severity of COVID-19 [22].
FIG 1
Coverage plot across hCoV-19/USA/CA-SMCPHL-072321.3/2021 (EPI_ISL_6159211) whole-genome
assembly. Coverage depth values were obtained by mapping Illumina reads to the reference
Wuhan-Hu-1 genome NC_045512.2 and expressed as the number of sequence reads at each
nucleotide position along the length of the CA-SMCPHL-072321.3 genome, which shows
a deletion in the ORF7a gene (around 27.5 Kbp). Mean sequencing depth was 5,066 reads.
FIG 1
Previous studies reported complete ORF7a gene loss [23, 24], multiple length in-frame
and frame-shift deletions in ORF7a 25, 26, 27, 28, 29, 30, 31, as well as large ORF7a∆370,
ORF7a∆227, and ORF7a∆392 that resulted in the fusion of ORF7a with ORF6, ORF7b, and
ORF8 genes, respectively [32, 33]. However, the ORF7a∆336 reported here is currently
the longest detected in-frame deletion within the ORF7a gene, which makes the gene
only 30 nt long (Figure 2
). ORF7a deletions are found to impact SARS-CoV-2 pathogenicity [31]. In vitro viral
challenge experiments demonstrated that the C-terminal truncation of ORF7a results
in a replication defect, which was found to be associated with elevated IFN response
to SARS-CoV-2 [31]. It was also shown that complete deletion of ORF7a reduces viral
replication [34]. This suggests that strains with deletions in ORF7a are more likely
to emerge in immunocompromised patients. Hence, further experiments are needed to
determine the functional outcomes of different deletions.
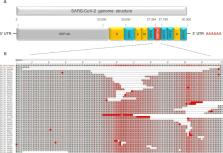
FIG 2
Complete SARS-CoV-2 genome organization diagram (A) and alignment of ORF7a gene sequences
(B). To avoid sequences with gaps due to poor sequencing coverage, only sequences
containing truncations and deletions in the ORF7a gene, that had high sequencing coverage,
were downloaded from GISAID (Table S1). The multiple amino acid alignments of the
ORF7a gene sequences (n = 47) with reference genome Wuhan-Hu-1 NC_045512.2 were performed
by MUSCLE algorithm in MEGA software v. 7.0.26. Frequency-based differences coloring
of aligned ORF7a proteins were visualized in NCBI MSA Viewer v. 1.20.1. The GISAID
accession number of the hCoV-19/USA/CA-SMCPHL-072321.3/2021 (EPI_ISL_6159211) from
this study is shown in red. The 336-nt (112-amino acid) deletion in CA-SMCPHL-072321.3
ORF7a was the longest observed deletion within the ORF7a gene sequences available
from GISAID and GenBank as of October 2021.
FIG 2
Unfortunately, a variety of deletions in the ORF7a region can be under-investigated
similarly to ORF8 deletions that have been shown to often not be reported by the standard
data analysis pipelines, which are frequently simply represented by stretch ambiguous
bases or as gaps in the consensus sequence due to poor NGS coverage [35]. Thus, non-canonical
genes are generally excluded from genomic and clinical analyses despite their importance
for understanding SARS-CoV-2 evolution and replication dynamics, which have vital
implications in vaccine development and control strategies for COVID-19 [6, 36, 37].
These findings highlight the necessity of submission of the raw sequencing reads in
public databases in order to assess the spread of deletion strains.
FUNDING
This work was supported by the CDC ELC CARES and Cooperative Agreement #NU60OE000103.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal
relationships that could have appeared to influence the work reported in this paper
Related collections
Most cited references33
- Record: found
- Abstract: found
- Article: not found
Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China
Chaolin Huang, Yeming Wang, Xingwang Li … (2020)

- Record: found
- Abstract: found
- Article: found
A new coronavirus associated with human respiratory disease in China

- Record: found
- Abstract: found
- Article: found
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation
Daniel Wrapp, Nianshuang Wang, Kizzmekia S. Corbett … (2020)