- Record: found
- Abstract: found
- Article: found
Myofibroblast differentiation and its functional properties are inhibited by nicotine and e-cigarette via mitochondrial OXPHOS complex III
Read this article at
Abstract
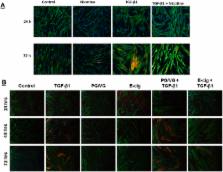
Nicotine is the major stimulant in tobacco products including e-cigarettes. Fibroblast to myofibroblast differentiation is a key process during wound healing and is dysregulated in lung diseases. The role of nicotine and e-cigarette derived nicotine on cellular functions including profibrotic response and other functional aspects is not known. We hypothesized that nicotine and e-cigarettes affect myofibroblast differentiation, gel contraction, and wound healing via mitochondria stress through nicotinic receptor-dependent mechanisms. To test the hypothesis, we exposed human lung fibroblasts with various doses of nicotine and e-cigarette condensate and determined myofibroblast differentiation, mitochondrial oxidative phosphorylation (OXPHOS), wound healing, and gel contraction at different time points. We found that both nicotine and e-cigarette inhibit myofibroblast differentiation as shown by smooth muscle actin and collagen type I, alpha 1 abundance. Nicotine and e-cigarette inhibited OXPHOS complex III accompanied by increased MitoROS, and this effect was augmented by complex III inhibitor antimycin A. These mitochondrial associated effects by nicotine resulted in inhibition of myofibroblast differentiation. These effects were associated with inhibition of wound healing and gel contraction suggesting that nicotine is responsible for dysregulated repair during injurious responses. Thus, our data suggest that nicotine causes dysregulated repair by inhibition of myofibroblast differentiation via OXPHOS pathway.
Related collections
Most cited references26
- Record: found
- Abstract: found
- Article: not found
Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling.
- Record: found
- Abstract: found
- Article: not found
Electronic cigarette inhalation alters innate immunity and airway cytokines while increasing the virulence of colonizing bacteria.
- Record: found
- Abstract: found
- Article: not found