- Record: found
- Abstract: found
- Article: found
C-C Chemokine Receptor 2 (CCR2) Regulates the Hepatic Recruitment of Myeloid Cells That Promote Obesity-Induced Hepatic Steatosis
Read this article at
Abstract
OBJECTIVE
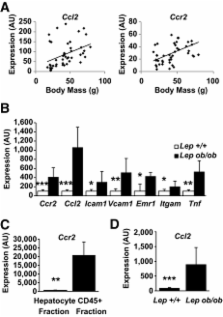
Obesity induces a program of systemic inflammation that is implicated in the development of many of its clinical sequelae. Hepatic inflammation is a feature of obesity-induced liver disease, and our previous studies demonstrated reduced hepatic steatosis in obese mice deficient in the C-C chemokine receptor 2 (CCR2) that regulates myeloid cell recruitment. This suggests that a myeloid cell population is recruited to the liver in obesity and contributes to nonalcoholic fatty liver disease.
RESEARCH DESIGN AND METHODS
We used fluorescence-activated cell sorting to measure hepatic leukocyte populations in genetic and diet forms of murine obesity. We characterized in vivo models that increase and decrease an obesity-regulated CCR2-expressing population of hepatic leukocytes. Finally, using an in vitro co-culture system, we measured the ability of these cells to modulate a hepatocyte program of lipid metabolism.
RESULTS
We demonstrate that obesity activates hepatocyte expression of C-C chemokine ligand 2 (CCL2/MCP-1) leading to hepatic recruitment of CCR2 + myeloid cells that promote hepatosteatosis. The quantity of these cells correlates with body mass and in obese mice represents the second largest immune cell population in the liver. Hepatic expression of CCL2 increases their recruitment and in the presence of dietary fat induces hepatosteatosis. These cells activate hepatic transcription of genes responsible for fatty acid esterification and steatosis.
Related collections
Most cited references24
- Record: found
- Abstract: found
- Article: not found
MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity.
- Record: found
- Abstract: found
- Article: not found
Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance.
- Record: found
- Abstract: found
- Article: not found