- Record: found
- Abstract: found
- Article: found
Improved chromosome-level genome assembly and annotation of the seagrass, Zostera marina (eelgrass)

Read this article at
Abstract
Background: Seagrasses (Alismatales) are the only fully marine angiosperms. Zostera marina (eelgrass) plays a crucial role in the functioning of coastal marine ecosystems and global carbon sequestration. It is the most widely studied seagrass and has become a marine model system for exploring adaptation under rapid climate change. The original draft genome (v.1.0) of the seagrass Z. marina (L.) was based on a combination of Illumina mate-pair libraries and fosmid-ends. A total of 25.55 Gb of Illumina and 0.14 Gb of Sanger sequence was obtained representing 47.7× genomic coverage. The assembly resulted in ~2000 unordered scaffolds (L50 of 486 Kb), a final genome assembly size of 203MB, 20,450 protein coding genes and 63% TE content. Here, we present an upgraded chromosome-scale genome assembly and compare v.1.0 and the new v.3.1, reconfirming previous results from Olsen et al. (2016), as well as pointing out new findings.
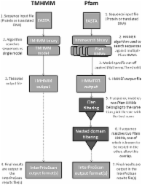
Methods: The same high molecular weight DNA used in the original sequencing of the Finnish clone was used. A high-quality reference genome was assembled with the MECAT assembly pipeline combining PacBio long-read sequencing and Hi-C scaffolding.
Results: In total, 75.97 Gb PacBio data was produced. The final assembly comprises six pseudo-chromosomes and 304 unanchored scaffolds with a total length of 260.5Mb and an N50 of 34.6 MB, showing high contiguity and few gaps (~0.5%). 21,483 protein-encoding genes are annotated in this assembly, of which 20,665 (96.2%) obtained at least one functional assignment based on similarity to known proteins.
Conclusions: As an important marine angiosperm, the improved Z. marina genome assembly will further assist evolutionary, ecological, and comparative genomics at the chromosome level. The new genome assembly will further our understanding into the structural and physiological adaptations from land to marine life.
Related collections
Most cited references50
- Record: found
- Abstract: found
- Article: not found
HISAT: a fast spliced aligner with low memory requirements.
- Record: found
- Abstract: found
- Article: not found
The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data.
- Record: found
- Abstract: found
- Article: found