- Record: found
- Abstract: found
- Article: found
A Case of a Newborn With Nemaline Myopathy From Al-Qunfudhah City, Saudi Arabia
Read this article at
Abstract
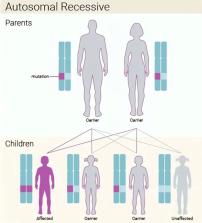
Nemaline myopathy is a primary skeletal muscle disorder and one of the congenital myopathies. It can be caused by mutations in at least 12 genes, with the nebulin ( NEB) gene being the most common. Here, we present the first case of a neonate with nemaline myopathy from Al-Qunfudhah, Saudi Arabia. A full-term baby boy was delivered via cesarean section due to decreased fetal movement. The baby was covered with a thick meconium stain. He was born with severe distress and underwent an endotracheal tube placement. The baby presented generalized muscle weakness, hypotonia, and areflexia. Examination revealed arthrogryposis, bilateral small chin, undescended testicle, joint deformity, hip dislocation, and clubfoot. Chest examination revealed conducting sound and bilateral equal air entry. Moreover, he experienced bilateral chest wheeze and conducting sound. All laboratory tests were normal, and whole-exome sequencing revealed pathogenic homozygous splice acceptor variant NEB gene c.8889+1G˃A. The patient was first suspected to have spinal muscular atrophy as there was no previous nemaline myopathy case reported from Al-Qunfudhah. However, the typical symptoms and genetic sequencing confirmed his condition. As the society in Al-Qunfudhah is known for consanguinity, as in our case, clinicians should identify other types of myopathy as it is expected to occur in further cases.
Related collections
Most cited references14
- Record: found
- Abstract: found
- Article: found
Spinal muscular atrophy
- Record: found
- Abstract: found
- Article: not found
Rare-disease genetics in the era of next-generation sequencing: discovery to translation.
- Record: found
- Abstract: found
- Article: not found