- Record: found
- Abstract: found
- Article: found
LGI1-Antibody Associated Autoimmune Encephalitis Complicated by Primary Polydipsia
letter

Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Dear Editor
Leucine-rich glioma-inactivated 1 (LGI1) antibody encephalitis is one of the subtypes
of the autoimmune encephalitis characterized by antibodies against the LGI1 region
of the voltage-gated potassium channels.[1] This subtype is characterized by behavioral
abnormalities, memory deficits, and classical facio-brachial dystonic seizures besides
hyponatremia. We hereby present a typical case of LGI1 associated encephalitis complicated
by primary polydipsia previously not reported in the literature.
A 28-year-old female was admitted to our department with complaints of memory impairment,
irrelevant talking, and abnormal body movements. As reported by her family members,
one month back just one week after her engagement, she started questioning about the
Mehendi applied on her hands and feet and could not recollect much about her engagement
ceremony. Her family members were also concerned about her not recognizing them properly
and asking repeatedly for meals even though she would have eaten already. Over the
past three weeks she developed abnormal movements of her right arm and face that would
last for few seconds and come repeatedly throughout day and night. On examination
she was confused, talking irrelevantly and there was no focal neurological deficit.
She had multiple episodes of facio-brachial seizures that would last for few seconds
only and come repeatedly after a few minutes. Her baseline investigations were normal
except for hyponatremia. Because of a subacute encephalopathy, memory deficits, characteristic
facio-brachial seizures, and hyponatremia, a provisional diagnosis of LGI1 encephalitis
were made. Her spinal fluid analysis revealed 5 cells/μL (all neutrophils) with normal
protein (45 mg/dl) and sugar (75 mg/dl with blood sugar of 96 mg/dl). Her MRI brain
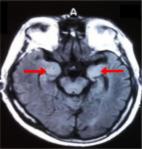
revealed bilateral symmetrical T2/FLAIR hyperintensities [Figure 1] in mesial temporal
lobes and insular cortices with no diffusion restriction or contrast enhancement.
Her CSF autoimmune panel was positive for the LGI1 antibody. She was managed with
pulse steroid therapy and antiepileptic drugs. However, she did not show any improvement,
and instead, she developed polyuria (average 9L/day) and polydipsia (average 8L/day)
without polyphagia. Her blood sugar was normal (88 mg/dl) and the main differentials
for polyuria and polydipsia were primary polydipsia and diabetes insipidus. However,
her serum sodium was persistently low and the water deprivation test did not show
a rise in serum osmolality ruling out diabetes insipidus [Table 1]. Hence a diagnosis
of LGI1-antibody autoimmune encephalitis complicated by primary polydipsia was made.
She was treated by IVIG over 5 days and she showed drastic improvement in encephalopathy,
facio-brachial seizures, and osmotic symptoms. She was discharged on steroids and
azathioprine and is currently on our follow-up.
Figure 1
FLAIR MRI sequence showing hyperintensities in bilateral medial temporal lobes
Table 1
Water Deprivation Test
Time
Urine Osmolality
Serum Sodium
At 0 hours
130 mOSm/Kg
127 meq/L
At 2 hours
277 mOSm/Kg
132 meq/L
At 4 hours
397 mOSm/Kg
125 meq/L
Limbic encephalitis associated with LGI1 antibodies has certain characteristic features
that may make clinical diagnosis easy. Facio-brachial seizures are one such feature;
these are dystonic movements of the face and ipsilateral arm that may remain for a
few seconds and are usually unilateral but can sometimes involve both sides.[2
3] Prominent psychiatric features like odd behavior, disinhibition, acute psychosis,
and memory impairment are other features of this disease.[4] Another characteristic
feature of LGI1 associated autoimmune encephalitis is hyponatremia that may be resistant
to treatment.[5] Hyponatremia may be seen in 60-80% of patients with LGI1 associated
encephalitis and is usually due to SIADH.[6] Imaging may be normal in up to 50% of
patients but may reveal T2/FLAIR hyperintensities in medial temporal lobes that are
usually unilateral but may also be bilateral.[7] First-line treatment in LGI1 encephalitis
is steroids usually in conjunction with intravenous immunoglobulin and/or plasmapheresis.
This therapy is usually effective in 80% of patients although the benefit may take
time to appear. Our patient had all the characteristic clinical features of LGI1-antibody-associated
encephalitis making diagnosis easier further strengthened by a positive CSF LGI1 antibody
test. Our patient however did not respond to initial steroid pulse therapy and developed
significant polyuria and polydipsia on the 8th day of admission. On the evaluation
of these osmotic symptoms, she had persistent hyponatremia with low urine and serum
osmolality. The water deprivation test did not increase serum osmolality although
there was an increase in urine osmolality thus confirming the diagnosis of primary
polydipsia.
Primary polydipsia is a condition characterized by excessive consumption of water
leading to dilute urine and hyponatremia. Primarily found in psychiatric disease patients
like schizophrenics, this disorder can also occur in patients with an organic brain
disease like sarcoidosis. This is the first case report of primary polydipsia in a
patient with autoimmune encephalitis published in the literature. Our patient was
managed with IVIG after which she showed a dramatic response in her seizures and polyuria
and polydipsia. As more and more cases of LGI1 encephalitis are reported worldwide
newer features of this disease become evident. Primary polydipsia is one such feature
that has not been previously reported.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms.
In the form the patient(s) has/have given his/her/their consent for his/her/their
images and other clinical information to be reported in the journal. The patients
understand that their names and initials will not be published and due efforts will
be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Related collections
Most cited references7
- Record: found
- Abstract: found
- Article: not found
Anti-LGI1 encephalitis: Clinical syndrome and long-term follow-up.
Agnes van Sonderen, Roland Thijs, Elias Coenders … (2016)
- Record: found
- Abstract: found
- Article: found
Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype.
- Record: found
- Abstract: found
- Article: found
Clinical features of limbic encephalitis with LGI1 antibody
Meiling Wang, Xiaoyu Cao, Qingxin Liu … (2017)